



A clinical trial is a culmination of the several stages of a drug or medical device development program that begins with the discovery of a candidate molecule followed by preclinical toxicology studies in ex vivo, in vitro, and animal models. Once the candidate molecule shows promising results in these stages, the next step involves clinical studies on human subjects. Drug testing in humans is often the most lengthy and expensive phase of the drug development timeline, and therefore requires extensive effort and careful execution to maximize the candidate’s chances of success. In addition to scientific evaluation, clinical studies require approval by the United States Food and Drug Administration (US FDA), the regulatory authority in the United States to administer the experimental drug in humans as well as ship it across state lines. This approval comes in the form of an Investigational New Drug (IND FDA) application that is required to be submitted by sponsors, investigators, or research institutes to the FDA to commence studies on human participants. The following figure shows the various stages of the drug development program (Figure 1) marking IND submission on the timeline.
The US FDA submission process is a critical step to ensure the safety and efficacy of new drugs. To ensure compliance with the FDA, drug developers must have an effective data strategy in place. This includes understanding the IND data requirements and having the plan to collect, analyze, and submit all relevant information to the FDA. With a well-thought-out drug development strategy in USA, drug developers can maximize their chances of success when submitting their new drugs for review.
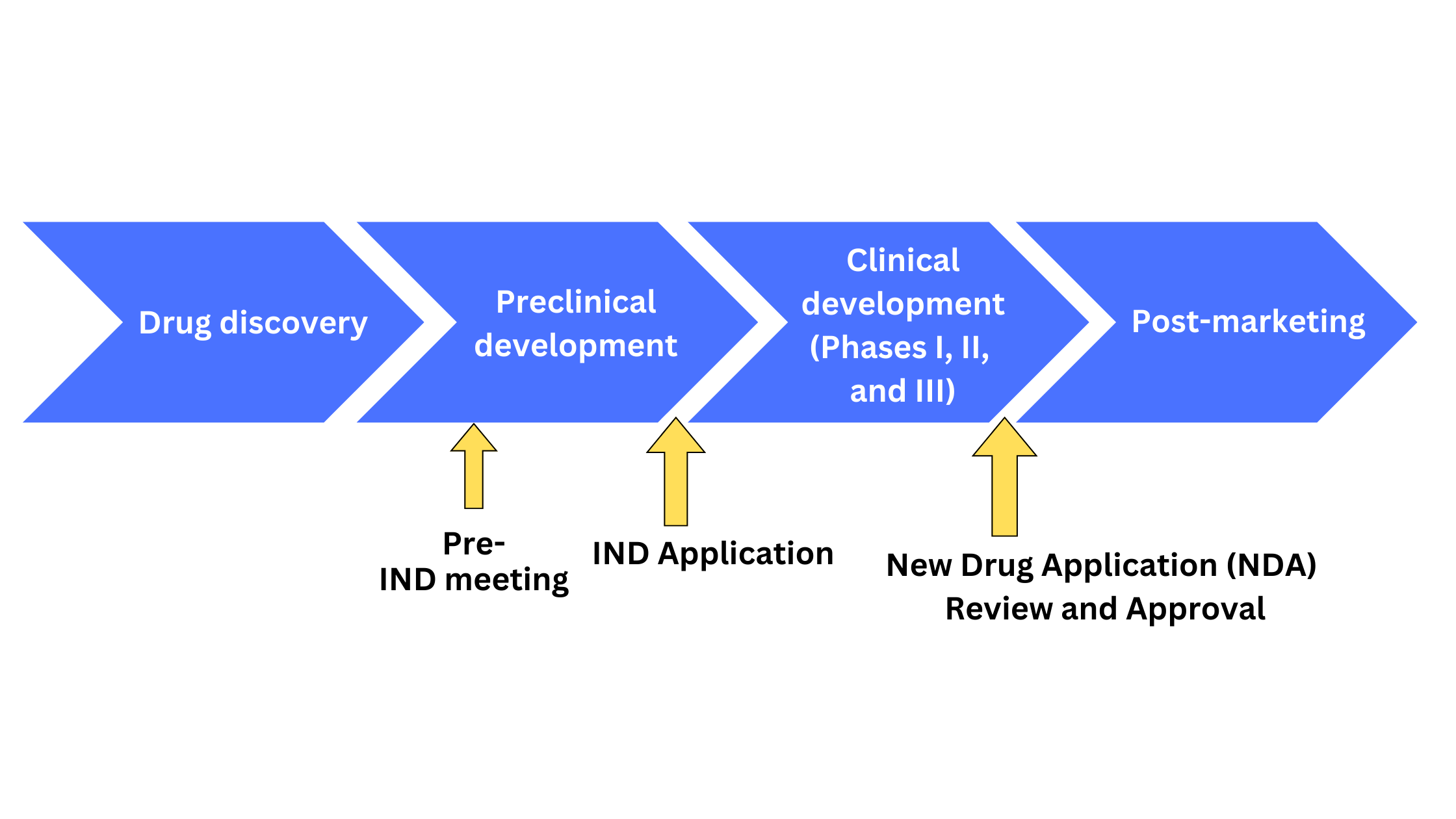 Figure 1. Schematic representation of drug development stages
Figure 1. Schematic representation of drug development stages
Why is an IND required?
The IND is a comprehensive document that contains all the information gained from preclinical and other studies in an organized format. The FDA reviews and makes the decision to support further clinical studies from information in the IND that ultimately forms the basis of marketing approval. INDs can be submitted at any phase during clinical development to protect the safety and rights of subjects (Phase I) and to assure adequate scientific evaluation of the drug’s effectiveness and safety (Phase II and III). The Code of Federal Regulations CFR Title 21. Part 312 Investigational New Drug Application contains information on INDs as well as their content and format and should be reviewed thoroughly by sponsors or investigators prior to submission of an IND application.
An Investigational New Drug (IND) application is an essential part of the process for conducting a Phase 1 clinical trial in the United States. The IND is required by the FDA before any investigational drug can be tested on humans. It ensures that the drug has been properly tested, manufactured, and labeled for safety and efficacy before it can be used in a clinical trial. The IND also contains detailed information about the proposed study design, investigator qualifications, and other necessary information to ensure that the trial will be conducted safely and ethically. By having an IND in place, sponsors of Phase 1 clinical trials in the USA can ensure that their trials will meet all regulatory requirements and provide valuable data to support further development of their drug or device.
There are two main types of INDs:
Commercial IND:
These are submitted by sponsors with the intent to commercialize the product at a later stage
Research IND:
These are submitted by investigators or research institutions that do not intend to commercialize the product
Emergency use IND:
Allows FDA to authorize the use of a drug in an emergency for which there is no comparable treatment and that does not follow the exact format for the submission of an IND according to 21 CFR Section 312
When do I need to submit an IND?
An IND should be submitted when planning to test a new drug in humans, when planning to use an approved drug for an indication that has not been in the approved labeling, when administered at a different dosage or by a different route of administration, and when administered to a different population (unless certain exemptions apply).
IND content and format
As of 2018, commercial use INDs must be submitted using the electronic Common Technical Document (eCTD) format for acceptability by the FDA. The eCTD attempts to harmonize marketing application structure and format in the US, EU, Japan, and other countries adhering to International Council for Harmonisation (ICH) guidelines and helps to save on paper and storage resources for documents.
INDs must contain information from three main areas: animal pharmacology and toxicology, manufacturing information, and clinical protocols and investigator information (Figure 2).
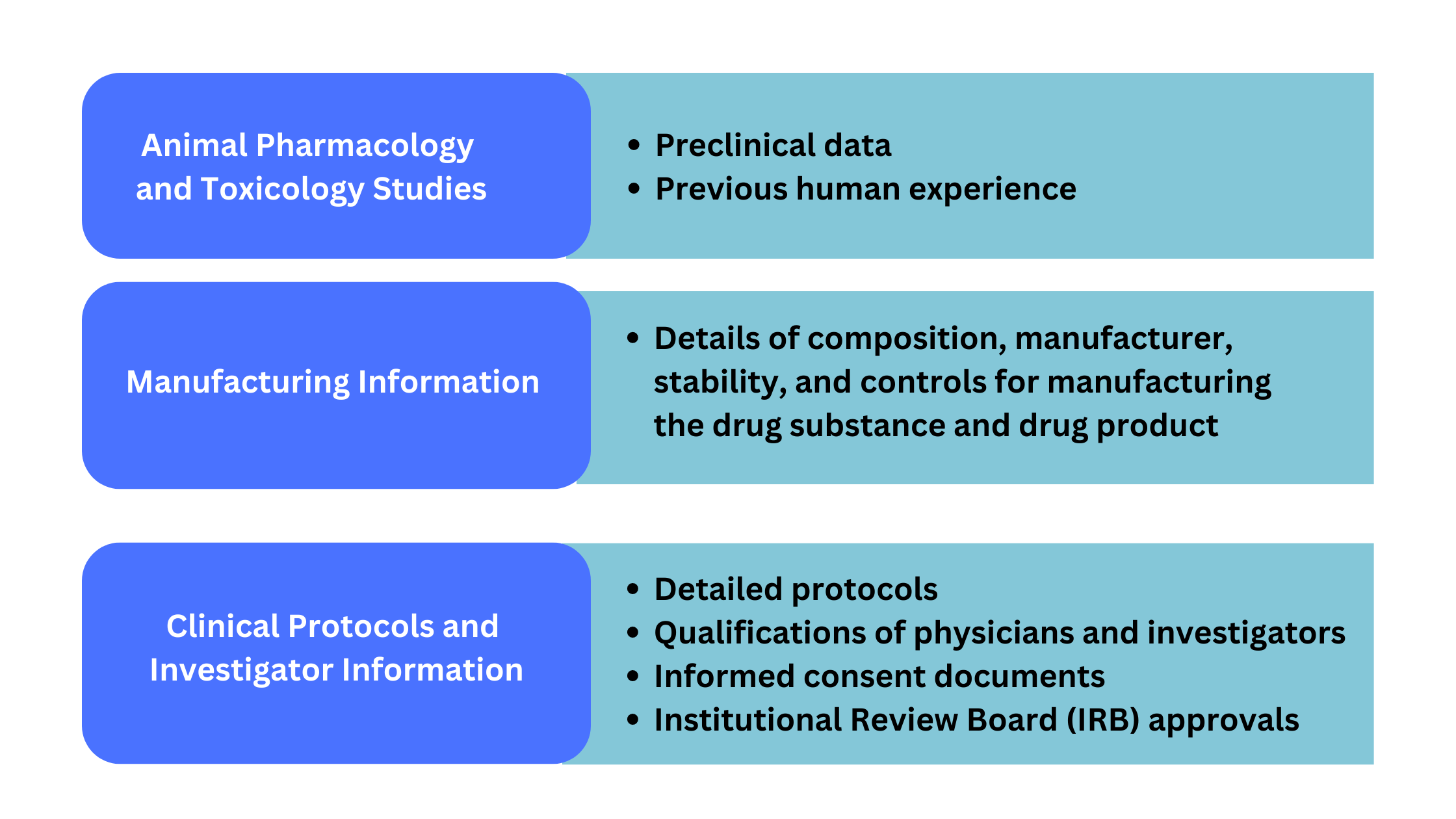 Figure 2. Information required for IND application
Figure 2. Information required for IND application
The eCTD format for the IND application contains 5 modules with specific information in each (Figure 3).
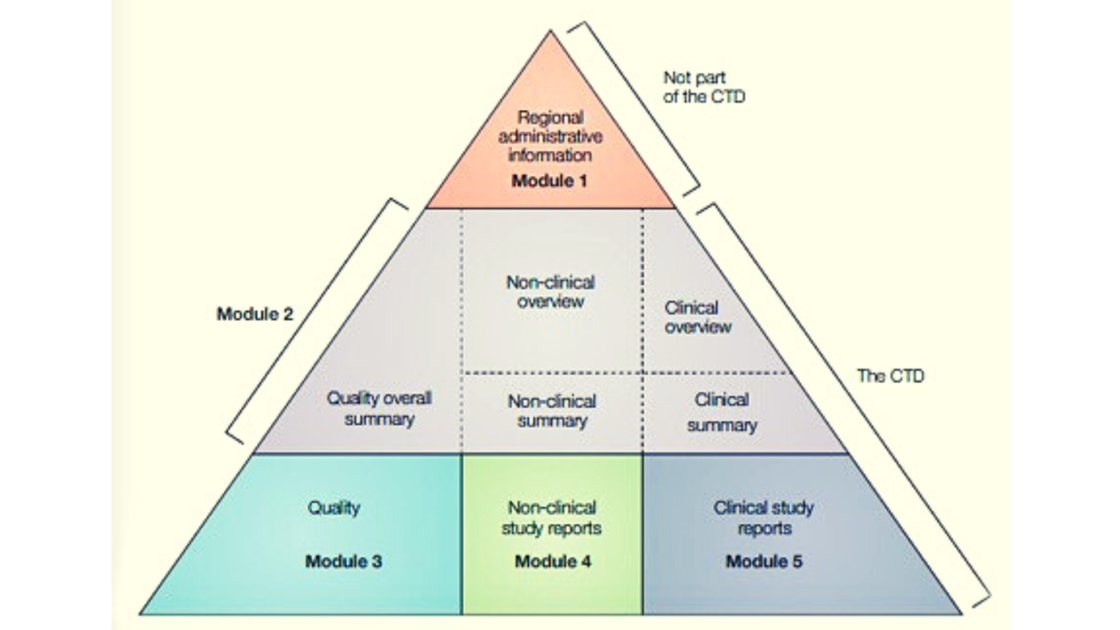
The information that sponsors/investigators must submit in an IND application based on 21CFR 312.23 can be arranged in the CTD format as follows and should contain:
Module 1-Regional and administrative information
- Cover letter (FDA 1571)-sponsor details, phase of clinical trial, details about clinical trial investigators and monitors, contract research organization (CRO) details, IRB information
- Table of contents-introductory statement and general investigational plan, along with information on previous human experience with the drug, investigational marketing experience and withdrawal information (if applicable) and plan for investigation for one year
- FDA 3674-certification of compliance with requirements ClinicalTrials.gov data bank
- Labeling-a copy of all labels and labeling to be provided to each investigator
- General Investigational Plan-contains information about the drug such as structural formula, pharmacological class, dosage form and dose, route of administration, and duration of trial
- Investigator’s brochure-contains information on the drug substance and formula, summary of pharmacological and toxicological effects in animals and humans (if known), summary of pharmacokinetics and biological disposition in animals and humans (if studies), safety and effectiveness in humans from prior clinical studies, possible risks, side effects, and precautions or special monitoring as per expected adverse events based on prior experience with the drug or similar drugs
Module 2-Summaries
- Quality overall summary-this includes information about the drug substance such as general information (nomenclature, structural formula), manufacture, characterization, control of drug substance, reference standards or materials, container-closure, and stability. Drug product data must contain description, pharmaceutical development, manufacture, control of excipients and drug product, reference standard or material, container-closure, and stability.
- Nonclinical overview-the aim of this section is to present data to ensure the safe use of the experimental drug in humans taking the pharmacological, pharmacokinetic, and toxicological studies into consideration.
- Pharmacology and pharmacokinetic studies: include pharmacokinetic, toxicokinetic, and metabolism studies with the methods used and analytical methods. Interspecies comparisons of metabolism and pharmacokinetic parameters (AUC, Cmax), and limitations for extrapolation of animal data to humans should be discussed.
- Toxicology studies: onset, severity, and duration of toxic effects, dose-dependency, reversibility (or irreversibility), and species and gender differences. Genotoxicity, carcinogenicity, toxic signs, causes of death, pathologic findings, pre- and postnatal toxicity, safety of drug during pregnancy and lactation, and local tolerance. Information Type of animal species, number of animals, route of administration and dosages, duration of treatment, systemic exposure should be included.
- Nonclinical summary-this includes written and tabulated summaries for pharmacology, pharmacokinetic, and toxicology studies
- Pharmacology written summary and tabulated summary-primary pharmacodynamics studies with other drugs in the class, secondary pharmacodynamic studies summarized by organ systems, safety pharmacology studies, pharmacodynamic drug interactions. The tabulated summary should contain information organized into sections such as test system, method of administration, dose, species/strain, gender, and organ systems evaluated, etc.
- Pharmacokinetics written summary and tabulated summary-method and validation parameters for analytical procedure used for biological samples in the study, absorption studies (in vitro extents and rate, BA/BE studies), distribution studies (tissue distribution, protein binding, and placental transfer studies), metabolism studies (metabolic pathways, pre-systemic metabolism, P450 in vitro studies, enzyme induction and inhibition), excretion studies (route and extent, excretion in milk), pharmacokinetic drug interactions. The tabulated summary should include data under the following headings species, vehicle/formulation, gender, dose, method of administration, sample, analyte, assay, feeding condition, and sampling time(s).
- Toxicology written summary and tabulated summary-single-dose toxicity (species, route), repeated-dose toxicity (species, route, duration), genotoxicity (in vitro mammalian and non-mammalian system, in vivo mammalian system), carcinogenicity studies (long-term and short-term), reproductive and developmental toxicity, local tolerance, and other studies (immunogenicity, antigenicity, dependence, studies on impurities/metabolites). Tabulated summaries should include the species/strain, dose, route of administration, duration, observed maximum non-lethal dose, and any noteworthy findings.
- Clinical overview-includes the product development rationale (pharmacological class, target indication, current major therapies, summary of ongoing and planned clinical studies), biopharmaceutics (bioavailability issues related to formulations), clinical pharmacology (pharmacokinetics: comparative PK related to intrinsic and extrinsic factors, absorption, protein binding, metabolic pathways, pharmacodynamics: mechanism of action and receptor binding, PK/PD relationships, genetic differences, immunogenicity and clinical microbiology), efficacy data (relevant patient population, study design, end point(s), controls, statistical methods), safety (adverse effects and monitoring, animal toxicology, overdose reactions), benefits and risks.
- Clinical summary-This section generally includes a tabulated summary and comparison of individual studies related to biopharmaceutics and analytical methods, clinical pharmacology, clinical efficacy, and clinical safety.
Module 3-Quality
This module contains chemistry, manufacturing, and controls data for the drug substance and drug product.
- Drug substance-general information (name, manufacturer), nomenclature (CAS no., chemical name), structure (formula and stereochemistry), general properties (pH/pKa, solubility, melting point, hygroscopicity, polymorphic form), manufacturer’s name and address, description of manufacturing process and controls (schematic flow diagram of process, raw materials, catalysts, solvents, pH, critical steps, equipment, and operating conditions), control of raw materials and solvents, control of critical steps and intermediates with acceptance criteria process validation and evaluations, and manufacturing process equipment. Specification criteria for drug substance, analytical procedures and validation criteria, batches and batch analyzes, and specification, reference standards and materials, container-closure system description, and tabular listing of stability protocol and studies
- Drug product-description of drug product, dosage, composition, type of container system, drug substance, excipients, formulation development, results from in vitro/in vivo comparative studies, physicochemical and biological properties relevant to performance (pH, dissolution, particle size, flow properties, rheology, potency, polymorphism, biological activity, etc.), manufacturing process development (key validation parameters), container closure systems microbiologic attributes, compatibility, batch formula, description of manufacturing process and controls, process validation, excipient controls, stability data (stress testing, photostability tests)
Module 4-Nonclinical study reports
This section involves the organization of nonclinical study reports which include pharmacology (primary and secondary pharmacodynamics, safety pharmacology, and pharmacodynamic drug interactions), pharmacokinetics (analytical methods and validation reports, absorption, distribution, metabolism, excretion, pharmacokinetic drug interactions), toxicology (single-dose, repeat dose, genotoxicity, carcinogenicity, reproductive and developmental toxicity, local tolerance, and other toxicity studies such as antigenicity, immunogenicity, tolerance, immunotoxicity, etc.), and literature references.
Module 5-Clinical study reports
This section involves the organization of clinical study reports which include biopharmaceutic study reports (bioavailability study, comparative BA/BE studies, In vitro-in vivo correlations, bioanalytical and analytical methods), PK study reports (plasma protein binding, hepatic metabolism and drug interactions, human biomaterial studies), human PK studies (healthy subject and patient PK and tolerability reports, intrinsic and extrinsic factor PK reports, population PK reports), human PD reports (healthy subject and patient PK/PD reports), efficacy and safety reports (controlled and uncontrolled clinical study reports,), reports of post-marketing experience, case report forms and individual patient listings, and literature references.
IND Submission Process to the US FDA
An IND can be submitted by the sponsor/investigator at any phase of the clinical trial stage but must be submitted prior to testing the experimental drug in human participants. Figure 4 shows the steps for submission of the IND application to the FDA using the eCTD format for electronic submissions.
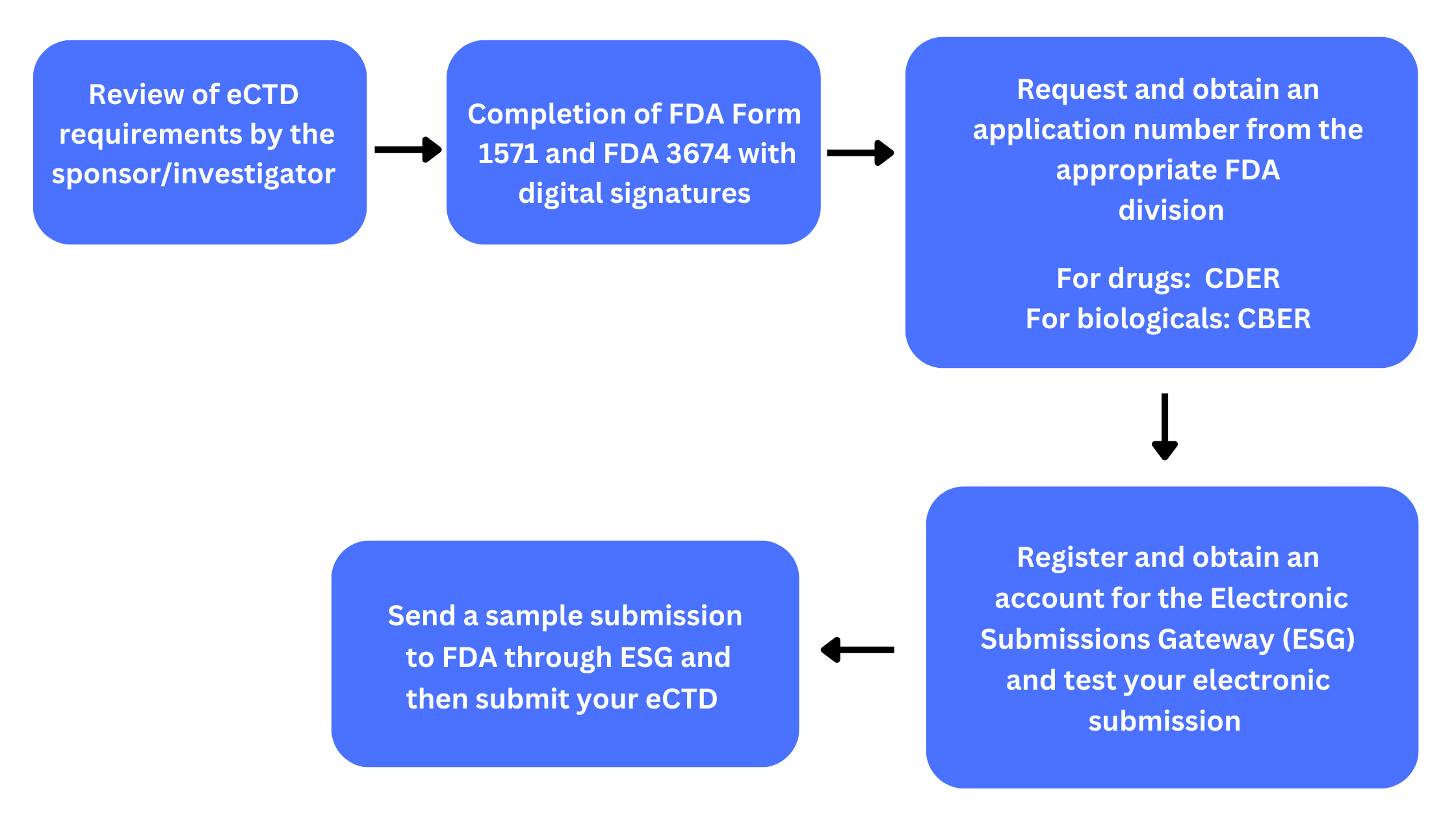
Figure 4. Submission process to FDA using the electronic system for INDs
Following receipt of the eCTD submission to the FDA, the FDA has a 30-day period to respond. The FDA can be in constant communication during this period to clarify any concerns or doubts about the study. If there is no response from the FDA during this window, the study can proceed. Otherwise, the FDA can place the study on partial hold or clinical hold which can be due to inadequate data in the submitted IND or safety concerns. It is the responsibility of the sponsor/investigator to address questions that the FDA has posed and respond to these in a separate submission to the FDA. The FDA can either lift the clinical hold allowing the clinical investigation to proceed, place the study on partial hold (with certain restrictions), or continue the hold.
In addition to submitting the initial IND, the sponsor/investigator must keep the IND current and submit any amendment to the IND such as protocol amendments, safety reports, and IND annual reports.
Thus, a thorough and clear understanding of the requirements for an IND especially in the eCTD format that is recommended by the FDA is necessary to maximize chances for a smooth transition of experimental drug to the clinical trial phase. Furthermore, reviewing the FDA submission process and maintaining contact with the FDA is essential in updating the IND and protecting patient safety in case of any adverse events or protocol deviations.
References
- Understanding FDA Regulatory Requirements for Investigational New Drug Applications for Sponsor-Investigators – PMC (nih.gov)
- M4E(R2): The CTD — Efficacy Guidance for Industry (fda.gov)
- Quality Overall Summary (QOS) in eCTD format (triphasepharmasolutions.com)
- Investigational New Drug (IND) Application | FDA
- Submit Using eCTD | FDA

