




Biological products have unlocked the potential for the management of several diseases such as cancers and autoimmune diseases for which treatment with small molecule, chemically synthesized drug molecules remain suboptimal. According to the Food and Drug Administration (FDA) biological products include vaccines, blood and blood components, allergenics, somatic cells, gene therapy, tissues, and recombinant therapeutic proteins. They are isolated from natural sources (human, animal, or microorganism) and may be produced by biotechnology or other advanced methods. The cost to produce and develop biologicals is high based on their complex structure and sophisticated technologies for production and analysis leading to considerable healthcare expenditures and as a result lower accessibility in most regions. This has prompted several countries to allow for the development of biological products that are demonstrated to be ‘high similar’ to an FDA-licensed biological product (or the reference product). These products are known as ‘biosimilars’ and are approved by the FDA via an abbreviated licensure pathway developed under the Biologics Price Competition and Innovation Act (BPCIA) and licensed under section 351(k) of the Public Health Service (PHS) Act. The term ‘biosimilarity’ means that the biological product is highly like the reference product notwithstanding minor differences in clinically inactive components and that there are no clinically meaningful differences between the biological product and reference product in terms of safety, purity, and potency of the product. As of January 2022, there are 33 FDA-approved biosimilar drugs with 21 available in the US market for treatment of various cancers, autoimmune diseases, and diabetes, and are expected to be 15-30% lower in cost compared to their reference products.
Small molecules vs. biologics and generics vs. biosimilars: what is the difference?
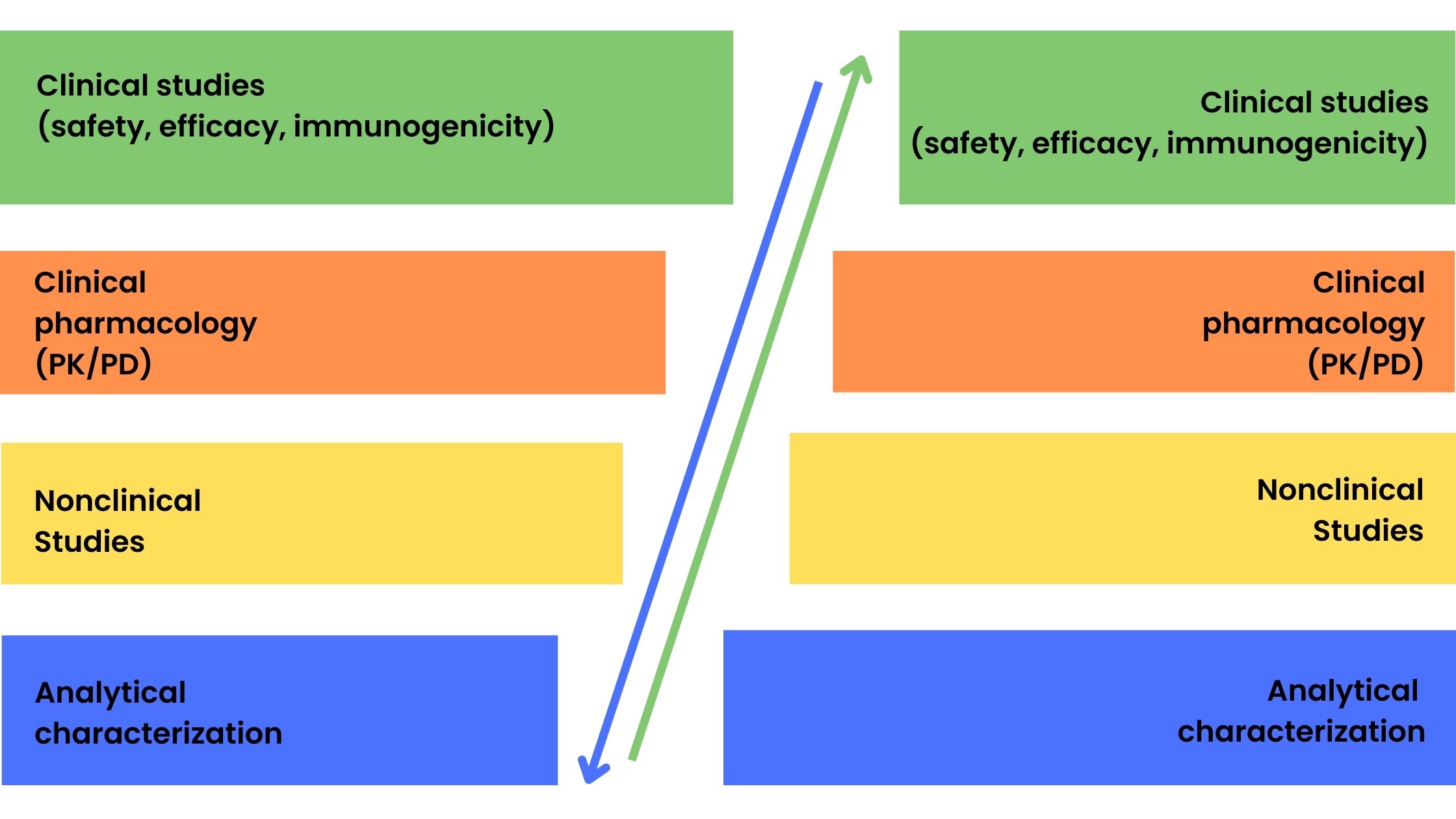
Generics are copies of small-molecule drugs and are chemically synthesized. The relatively simple synthesis process and well-defined and reproducible starting material allow for generics to be identical to the reference drug. However, this is not possible for biologicals that are large molecules with a complex chemical structure produced by genetic engineering and fermentation and purification processes. Therefore, the term biosimilar is used to denote ‘generic’ versions of biologicals. Generic versions of small-molecule conventional drugs are approved by the FDA using the Abbreviated New Drug Application (ANDA) pathway which involves the demonstration of bioequivalence to the reference molecule and usually does not require preclinical studies in animals and clinical trials in humans. In contrast, the greater variability between batches of biological products and minor differences in starting materials and manufacturing processes requires a full suite of tests from analytical characterization to clinical studies to be performed. Figure 1 shows the product development process for reference biologicals and biosimilar drugs. The stepwise approach to biosimilar drug development involves the development of a clinical program based on the degree of uncertainty identified after extensive analytical and structural characterization from previous stages of product development.
(a) (b)
Figure 1. Product development phases for reference biologicals (a) and biosimilar drugs (b)
Figure 1 also shows the difference in focus on certain areas of product development between reference biologicals and biosimilar drugs with a greater emphasis on clinical development for the innovators and more extensive analytical and quality studies for biosimilars. However, a totality-of-evidence approach is required for biosimilar approval which encompasses the following:
- Analytical characterization: physical, chemical, and biological activity to demonstrate that the biosimilar is comparable structurally and functionally to the reference product
- Nonclinical studies: demonstration that the biosimilar acts on the same target or physiological process and has the same toxicity profile as the reference using in vitro (cell culture) and in vivo (animal) models
- Clinical pharmacology and clinical studies: assessment of how the biosimilar is absorbed, distributed, metabolized, and excreted (PK) and how it works in the body including any safety and immunogenicity issues relative to the reference product
Clinical Trials for Biosimilars from a US FDA perspective
Although the demonstration of biosimilarity does require clinical studies, the nature and scope of studies required is dependent on the extent of uncertainty about biosimilarity obtained from analytical characterization and preclinical (animal) data. The US FDA guidance requires sponsors to perform human PK and PD studies, clinical immunogenicity studies, and in some cases comparative clinical studies for the biosimilar approval process (Figure 2).
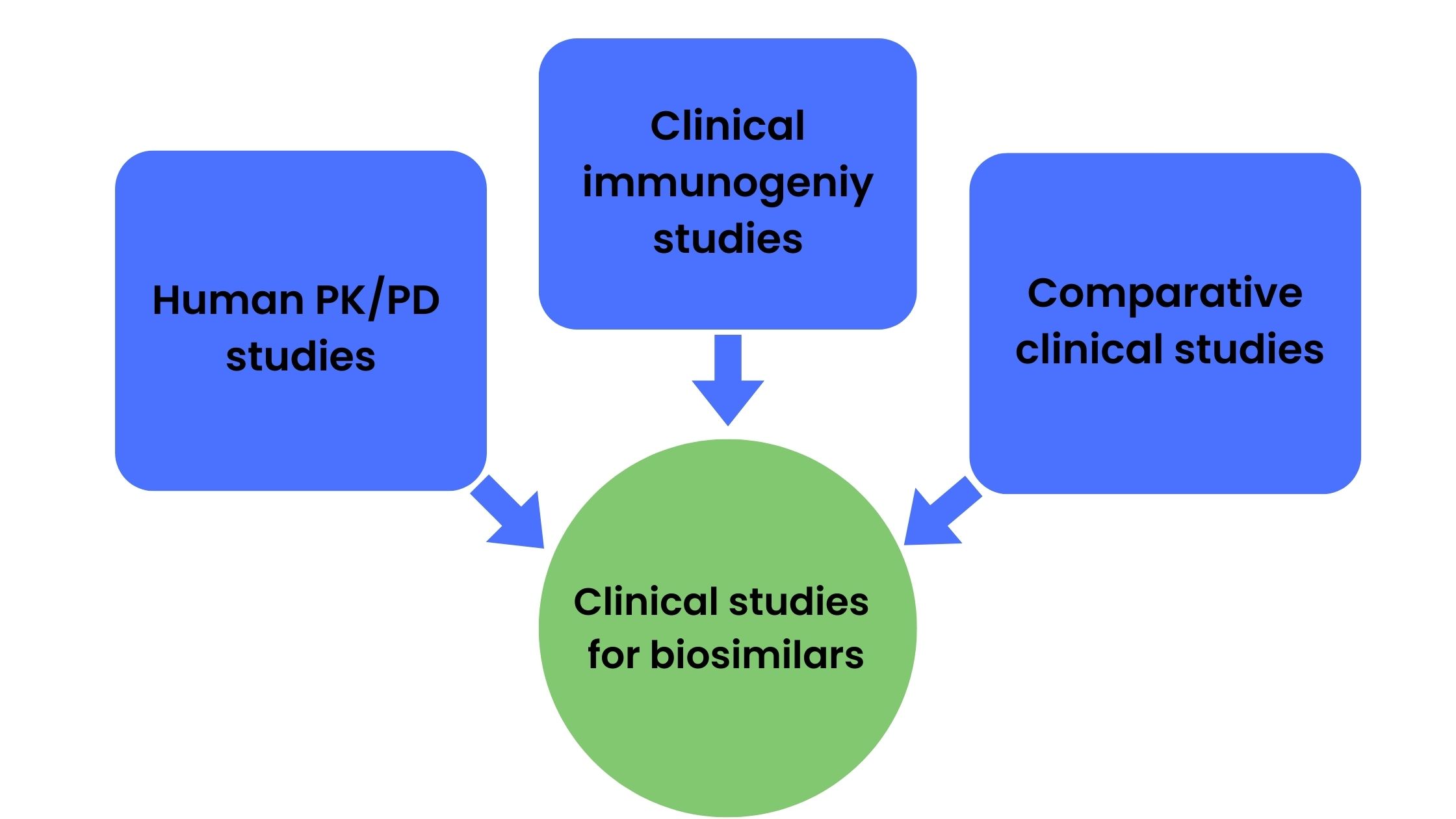
Figure 2. Studies commonly required for biosimilar approval process
Human PK and PD studies
These studies are conducted to demonstrate biosimilarity between the reference product and biosimilar drug and involve a comparison of serum plasma concentration-time profiles. PK/PD studies should be conducted in healthy subjects or patients using a route of administration or dose that is sensitive enough to allow for the detection of differences in PK and PD profiles. Pharmacodynamic (PD) parameters chosen should be linked to the clinical outcome and be sensitive enough to detect clinically meaningful differences. The establishment of similar PK, PD, and immunogenicity profiles is generally convincing enough to demonstrate the absence of clinically relevant differences between the products and involves similar dose-response and PK profiles.
Clinical immunogenicity studies
These studies are especially important for biologics to evaluate potential differences in immune responses which may affect the safety and effectiveness of the product by altering PK, inducing anaphylaxis, or promoting the development of neutralizing antibodies. The study design and extent of the immunogenicity study should be based on information gathered from previous studies such as analytical and nonclinical studies as well as the severity and consequences of the immune response. The selection of the study population and treatment regimen should also be sensitive enough to detect differences in response and immunogenicity studies of the reference product should be referred to when selecting immunogenicity or PD endpoints. Various antibody parameters such as titer. Specificity, the time course of development, persistence, etc. along with a suitable development and validation of immunogenicity assays should be done.
Comparative clinical studies
These studies only need to be performed if there is any residual uncertainty surrounding the issue of clinically meaningful differences between the reference biological and biosimilar based on analytical and structural characterization studies, preclinical studies, PK/PD evaluations, and clinical immunogenicity results. Various factors such as the complexity of reference biological, clinical experience with the proposed biological such as risk-benefit profile, appropriate endpoints and biomarkers, and the degree to which human PK/PD studies and differences in structure, function, and nonclinical studies can predict differences in clinical outcomes determine the type and extent comparative clinical studies required to demonstrate biosimilarity.
Considerations for clinical trials of biosimilars
Endpoints
PK and PD endpoints chosen should preferably correlate with clinical outcomes and should be relevant to the disease state while also being sensitive enough to detect differences between products that affect safety and efficacy. In most cases, primary endpoints used in clinical trials for the reference biological are used unless adequate justification is given for use of alternate endpoints.
Study population
The study population chosen may be healthy subjects or patients with a certain condition and should be sensitive enough to detect clinically meaningful differences between the reference biological and biosimilar. Certain subjects may be more prone to exhibiting adverse effects such as those on immunosuppressant therapy and therefore trials should be performed on them. In most cases, the study population chosen in the development of the licensed product is used for biosimilarity trials.
Biological product characteristics
The half-life of the biological is usually the deciding factor for the type of trial design to be used. For biologicals with a short half-life (< 5 days) and rapid PD response, a cross-over design is suitable whereas parallel trial designs are recommended for products with a long half-life. Other factors such as the dose and route of administration also need to be considered. The nature and complexity of the biological will also dictate the extent and depth of immunogenicity studies and safety monitoring that will be required during the trial.
Sample size
This is one of the most critical aspects of a biosimilarity trial to ensure that it is sufficiently powered to detect differences between the biosimilar and reference product. Equivalence margins and treatment effects dictate the minimum sample size required and should be factored in whilst calculating the sample size.
Duration of study
The duration of the study is guided by the condition for which the biological is to be used with longer studies necessary for chronic conditions to capture adequate endpoints related to efficacy and safety.
Study design
Since biosimilarity trials involve proving that the biosimilar is equivalent to the reference biological, equivalence and noninferiority trial designs are most suitable. Typically equivalence designs with symmetric inferiority and superiority margins should be used whereas asymmetric margins can be used in certain cases to rule out inferiority and allow for smaller sample sizes.
Statistical analyzes
In most cases, 80-125% acceptance range is used for PK and PD parameters such as maximum plasma concentration (Cmax) and area under the curve (AUC).
Biosimilars lower costs and provide access to biological treatments for diseases that are otherwise difficult to manage. Designing clinical trials that are clinically and statistically sound is imperative to increasing the approval of biosimilars thus benefiting payers, physicians, and patients. It is important for sponsors/investigators of biosimilars to understand and refer to US FDA guidelines on biosimilar drugs to facilitate and expedite approval.
References
- Scientific Considerations in Demonstrating Biosimilarity to a Reference Product | FDA
- Introductory Chapter: Biosimilars – A Regulatory and Clinical Perspective | IntechOpen
- FDA Webinar – Overview of the Regulatory Framework and FDA’s Guidance for the Development and Approval of Biosimilar and Interchangeable Products in the US | FDA
- What Are “Biologics” Questions and Answers | FDA
- Biosimilars in the EU – Information guide for healthcare professionals (europa.eu)
