The United States Food and Drug Administration (FDA) has strict regulations regarding pharmacovigilance activities that are a part of post-marketing studies for drugs and medical devices that are required as part of a New Drug Application (NDA). The Code of Federal Regulations Title 21 contains various sections related to pharmacovigilance, adverse event reporting, and drug safety and these regulations are governed and enforced by the FDA. Some of the relevant regulations for pharmacovigilance include:
Title 21 CFR Part 314
Which includes post-marketing and pharmacovigilance activities that are required in an NDA such as periodic reports, individual case safety reports (ICSR), and electronic reporting formats. This part includes several subparts that are related to risk evaluation and mitigations strategies (REMS), adverse event reporting, dispute resolution, and any deviations.
Title 21 CFR Part 600
This relates to reporting requirements for adverse events related to biological products, vaccines, and blood products.
The FDA has released a guidance document in 2005 on “Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment” that provides recommendations and best practices for safety signal evaluations. It includes information for the development of case series, data mining from databases such as FDA Adverse Event Reporting System (FAERS) and Vaccine Adverse Event Reporting System (VAERS), labelling modifications depending upon incidence rates of the safety signal, and data collection and signal collection through non-randomized, observational studies such as pharmacoepidemiologic studies, registries, and surveys. It also emphasizes the importance for the development of a pharmacovigilance plan and specifies what elements need to be included in it.
Figure 1 shows the various regulations that are required to be followed by sponsors/manufacturers for pharmacovigilance activities.
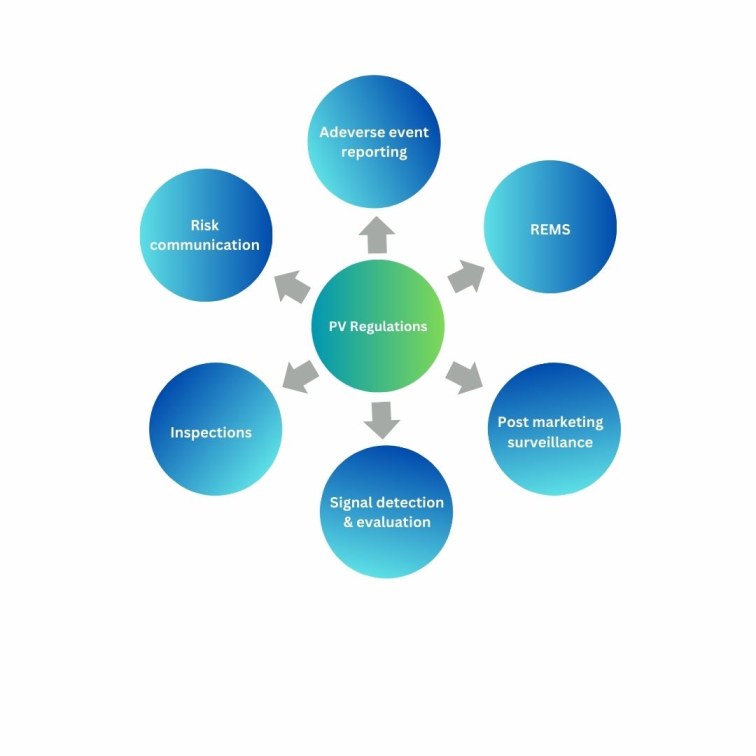 Adverse event reporting
Adverse event reporting
The FDA has the FAERS and VAERS databases for voluntary reporting of adverse events received from post-marketing studies, observational studies, or real-word evidence (RWE). These databases contain information reported by consumers or healthcare professionals and is used by the regulatory agency to make decisions regarding the use of the product or changes in labelling that may be needed.
Risk Evaluation and Mitigation Strategies (REMS)
The US FDA mandates the development of REMS programs for drugs with safety concerns to ensure that the benefits outweigh the risks. They are required to educate and inform healthcare professionals on safe prescribing information and are not intended to mitigate adverse effects but rather increase awareness about them.
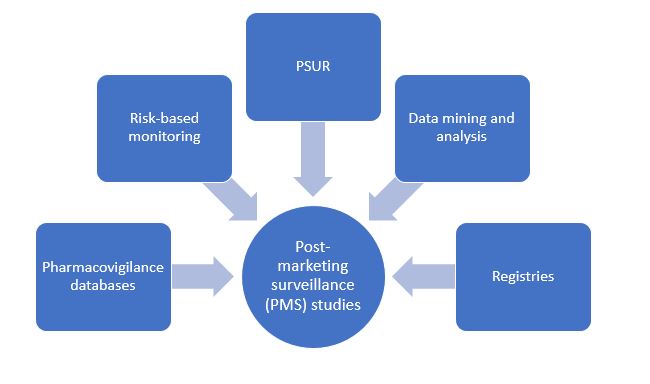
Post-marketing surveillance (PMS)
PMS studies are an essential component for FDA-regulated drugs and medical devices and for a part of the product lifecycle. FDA programs such as Med Watch help to collect unexpected or previously unobserved adverse events not captured during clinical trials. They help regulatory agencies make decisions regarding the fate of the product, such as withdrawals, limited usage, or change in labelling conditions.
Signal detection and evaluation
Potential safety issues that are entered on the FAERS database are evaluated by the FDA to characterize the risk and determine if there is a casual relationship between the event and the product. Once an issue has been deemed to present a safety concern, the FDA is responsible for taking appropriate action and communicating the risk to healthcare professionals such that they can modify their prescribing patterns.
Inspections
The FDA’s Post marketing Adverse Drug Experience (PADE) compliance program monitors the manufacture/sponsors compliance with post marketing laws and regulations for FDA-regulated products. Noncompliance and deviations result in warning letters and regulatory actions.
Risk communication
The FDA website contains a Drug Safety Communications page that provides patients and healthcare professionals with up-to-date safety information on regulated products which includes adverse events and long-term side effects.
In April 2022, the FDA released a guidance document on the submission of post-marketing safety reports in an electronic format. It describes the submission process and timeline for individual case safety reports (ICSRs) to the FAERS system through the electronic submissions gateway (ESG) or safety reporting portal (SRP). It describes the procedure that involves setting up an account and password, details on unique identification numbers and periodic submissions of ICSRs, label submissions for unapproved prescription and nonprescription products, and ICSR submissions and attachments. The guidance also describes the acknowledgement of submission by the FDA to the FAERS database and contact information in case of any problems with the submission process. Submission methods such as physical submissions and guidelines for these are stated in case of non-functionality of the FAERS system. The format and content of waiver requests, reasons for waiver, and portal for submission are covered in this guidance.
Another guidance document on the electronic transmission of ICSRs for drugs and biological products was released in August 2022. This guidance describes technical requirements for submitting ICSRs to the FAERS database as per ICH E2B(R3) specifications along with incorporation of FDA regional data elements. File structures, data elements, formats, coding structures, medical terminologies, and transmission standards are described in this guidance. Additionally, requirements for expedited reporting, follow-up of ICSRs, and contact information in case of misinformation is also specified. The overall aim of this guidance is to ensure accuracy, consistency, and streamline the process for submission of ICSRs to the FDA allowing for safeguarding of patient health and well-being.
Therefore, it is required that all manufacturers, sponsors/investigators, and companies adhere to FDA regulations and comply with necessary standards. The current set of regulations help collect adverse event information that can provide feedback on the safety of the product. As electronic reporting is becoming increasingly popular, most of the new FDA guidances focus on the submission and format for electronic submissions to the FDA database which can facilitate exchange of information within the U.S. and globally allowing for patient safety.
Read More– US FDA Foreign Clinical Trial Data Requirements
References
- FDA Regional Implementation Guide for E2B(R3) Electronic Transmission of Individual Case Safety Reports for Drug and Biological Products | FDA
- Providing Submissions in Electronic Format — Postmarketing Safety Reports | FDA
- CFR – Code of Federal Regulations Title 21 (fda.gov)
- 21 CFR 314.80 – Postmarketing reporting of adverse drug experiences. – Content Details – CFR-2011-title21-vol5-sec314-80 (govinfo.gov)