




The United States has always been and remains to be the leading place for the conduct of clinical trials. According to Clinicaltrials.gov, the largest clinical trials registry, 32% of registered clinical trials were conducted in the U.S. as of May 2022 (1). Factors such as the availability of qualified healthcare professionals, high-quality infrastructure and facilities, cutting-edge research, efficient regulatory system, and a high standard of ethics and participant protection make the U.S. the leading country for clinical trials.
Clinical trials follow extensive preclinical research to test the safety and efficacy of a new drug, medical device, or biological in humans. They are usually divided into three phases: phases I, II, and III which are designed to ascertain safety, pharmacokinetics, efficacy, dosage, and adverse events. Figure 1 shows the typical route from discovery and preclinical studies to the post-marketing phase (phase IV).
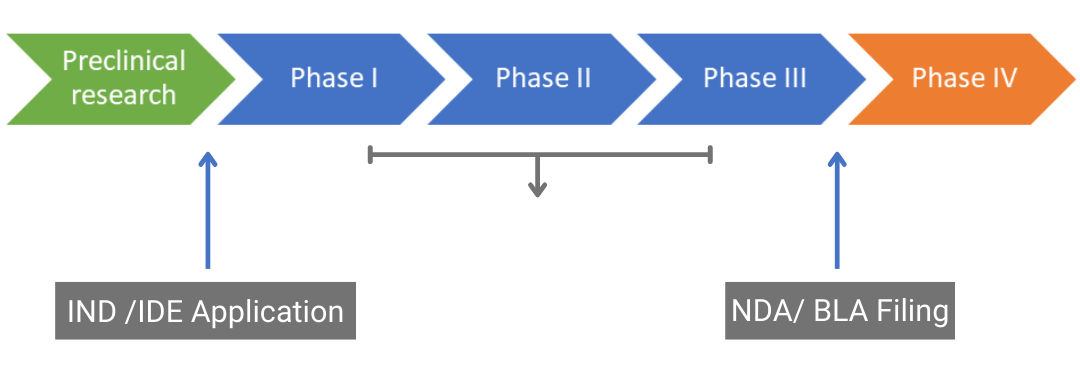
Drug approval pathway. IND: Investigational New Drug, IDE: Investigational Device Exemption, NDA: New Drug Application, BLA: Biologics License Application
Figure 1. Drug approval pathway. IND: Investigational New Drug, IDE: Investigational Device Exemption, NDA: New Drug Application, BLA: Biologics License Application
Clinical trials represent the longest and most expensive step in bringing drugs to the market and have the highest attrition rate, only 10% of drugs that enter phase I trials are granted marketing approval. Therefore, clinical trials should be conducted by experts that are
well-versed with all the regulations and guidelines in a particular region to boost the chances of drug approval.
The United States Food and Drug Administration (US FDA) is the regulatory body that approves and oversees the conduct of clinical trials for drugs, medical devices, and biologicals that are intended to be marketed in the U.S and is touted to have the most stringent standards for drug approval. The primary role of the FDA is to protect public health by ensuring that medicinal products and devices are safe and efficacious. Therefore, it is necessary for sponsors/investigators or contract research organizations (CRO) that are conducting clinical trials to be familiar with regulations and guidances that govern the conduct of clinical trials.
What you need to know to conduct a Clinical Trial in the United States
Clinical Trial in USA
1. Regulations
Sponsors, investigators, or CROs planning to carry out clinical trials in the U.S. need to comply with federal regulations that are listed in Part 21 Code of Federal Regulations (CFR). Details regarding these regulations are available on the official FDA website (https://www.fda.gov(2)). The applicable CFR regulations that are noteworthy include:
- 21 CFR Part 50: Protection of Human Subjects which related to informed consent from human subjects or legally authorized representatives. This part describes the elements of informed consent and exceptions.
- 21 CFR Part 54: Financial Disclosure by Clinical Investigators which details disclosure of financial agreements between the sponsor and clinical investigator in cases of financial interest of the investigator based on royalties or patents.
- 21 CFR Part 56: Institutional Review Boards provides details on the standards for the composition, operation, and responsibility of an IRB.
- 21 CFR 312: Investigational New Drug (IND) Application which includes procedures, requirements, and submission of INDs.
- 21 CFR 812: Investigational Device Exemptions is related to clinical investigations of devices.
- 21 CFR Part 814: Premarket Approval (PMA) of Medical Devices outlines the review process for approval or disapproval of PMAs
2. Good Clinical Practice (GCP) Guidelines
A thorough understanding of and compliance with Good Clinical Practice (GCP) guidelines by sponsors and investigators is essential in protecting the rights, confidentiality, and well-being of human subjects along with ensuring reliable and accurate clinical data. The U.S. uses the GCP guideline developed by the International Council for Harmonizationof Technical Requirements for Pharmaceuticals for Human Use (ICH). The ICH-GCP guideline includes the following information that should be followed by various parties involved in a clinical trial and the documentation required (2):
- Institutional Review Board (IRB)/Independent Ethics Committee (IEC): Responsibilities, composition, function, procedures, and records
- Investigator: Qualification and training, adequate staff availability and facilities, contracts with CROs, medical care of trial subjects, communication with IRB/IEC, compliance with the protocol, reporting of any protocol deviations, handling the investigational product (IP), obtaining informed consent, maintenance of source documentation, safety reporting
- Sponsor: Development of a quality management system using a risk-based approach, implementation and maintenance of quality control and quality assurance systems, oversight of any trial activities conducted by CROs, trial design, management, data handling, record keeping, investigator selection, financing, IRB/IEC approval confirmation, adequate nonclinical data to support the IP, suitable manufacturing and packaging conditions for the IP, supply and distribution of the IP, ongoing safety evaluation, and adverse drug reaction reporting, suitable monitoring, and audit plans
- Clinical trial protocol and protocol amendments: Trial design, objectives, selection and treatment of subjects, ethics, data handling and record keeping, statistics, assessment of safety and efficacy, financing, and insurance
- Investigator’s brochure contents and format
- Essential documents before, during, and after the trial
3. Regulatory pathways
Understanding the regulatory pathways for the approval of drugs, medical devices, and biologicals is important to expedite the approval process. The utilization of abbreviated routes for approval can benefit the sponsor by saving considerable time and money. The requirements vary for each pathway and need to be understood. The common pathways for approval that any sponsor should be familiar with are:
- New drugs: 505(b)(1) pathway for New Drug Application (NDA) which includes extensive clinical studies (phase I-III)
- Generic drugs: Abbreviated new drug application (ANDA) that emphasizes the importance of bioequivalence studies
- Repurposed drugs: 505(b)(2) pathway for repurposed or repositioned drugs which precludes the need for extensive preclinical and clinical studies. This pathway for regulatory approval focuses instead on compilation of existing information from FDA literature or labeling, and conduct of gap analysis and bridging studies
- Medical devices: Premarket notification (PMN) or 501(k) process for Class II devices that shows substantial equivalence to predicate devices. Premarket approval (PMA) and Investigational device exemption (IDE) for Class III medical devices
- In vitro diagnostic (IVD) devices: 510(k) or premarket approval (PMA) process depending on device classification
- Biologics: Traditional or full biologics license application (BLA) 351(a) for innovator biologicals and biosimilar BLA section 351(k) for biosimilar biologicals
4. Ethical considerations
Although ethics are a part of CFR and GCP guidelines, it is necessary to emphasize this aspect as the protection of human subjects is of utmost importance during a clinical trial. The three main principles for ethical research that are part of the ICH-GCP guidelines and which in turn are drawn from the Belmont report are respect for persons (informed consent), beneficence (benefits should outweigh risks), and justice (equitable selection and treatment of human subjects). It is imperative for the sponsor/investigator to obtain ethical approval from the IRB/IEC prior to commencing clinical research as well as voluntary consent from subjects or their legally authorized representatives (LARs). In addition to federal regulations, sponsors or CROs must be aware of state legislation such as protection of human subjects, confidentiality statements, rules related to consent from minors and prisoners, codes related to parenteral consent, etc. that may be required for conducting clinical trials in some states in the U.S.
5. Site requirements
Availability of resources at clinical trial sites, adequate drug storage facilities, and trained personnel are important when conducting a clinical trial. It is essential to have a clinical trial coordinator (CRC) at each trial site who is responsible for the day-to-day activities of the trial. Since documentation is a critical factor for trials and regulatory submissions, the site must have a system to store and archive all patient data records in a secure manner. Any equipment or instruments that are required for sample collection and processing must be available at the site in addition to suitable storage facilities such as cold storage to avoid sample degradation. Sites should also be chosen based on their proximity to hospitals or nursing homes to allow for recruitment of a sufficient number of subjects for the trial to meet enrollment targets.
Successful completion of a clinical trial in the USA requires the sponsor or CRO to have a strong knowledge of the requirements for clinical trials. This will help streamline the trial and regulatory approval process.
References
- Trends, Charts, and Maps – ClinicalTrials.gov
- E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1) Guidance for Industry, March 2018
