


Power medical device innovation through 510(k), PMA & De Novo Clinical Trials designed to satisfy FDA requirements, guidelines, and certification standards. From evidence generation to regulatory compliance, we enable confident FDA approval and clearance with precision, speed, and FDA Guidance, aligned execution.
510(k), PMA, and De Novo clinical trials are FDA regulatory pathways used to evaluate the safety and effectiveness of medical devices before they can be legally marketed in the United States. Each pathway has different clinical evidence requirements based on device risk and novelty.
510(k) Clinical Trials are typically required for moderate risk devices that can demonstrate substantial equivalence to an existing legally marketed device. Clinical studies may be needed to confirm safety, performance, and intended use when nonclinical testing alone is insufficient. Successful review results in FDA clearance.
PMA Clinical Trials apply to high risk, life sustaining, or implantable devices. These trials are more rigorous and usually involve pivotal clinical studies designed to provide valid scientific evidence of safety and effectiveness. Approval is granted only after comprehensive FDA review of clinical data, manufacturing controls, and quality systems.
De Novo Clinical Trials are intended for novel, low to moderate risk devices with no predicate device. Clinical studies help establish reasonable assurance of safety and effectiveness and create a new device classification. Once granted, De Novo clearance allows future devices to use this classification as a predicate.
Together, these clinical trial pathways ensure medical devices meet FDA requirements, follow regulatory guidelines, maintain compliance, and achieve clearance or approval based on robust clinical evidence.
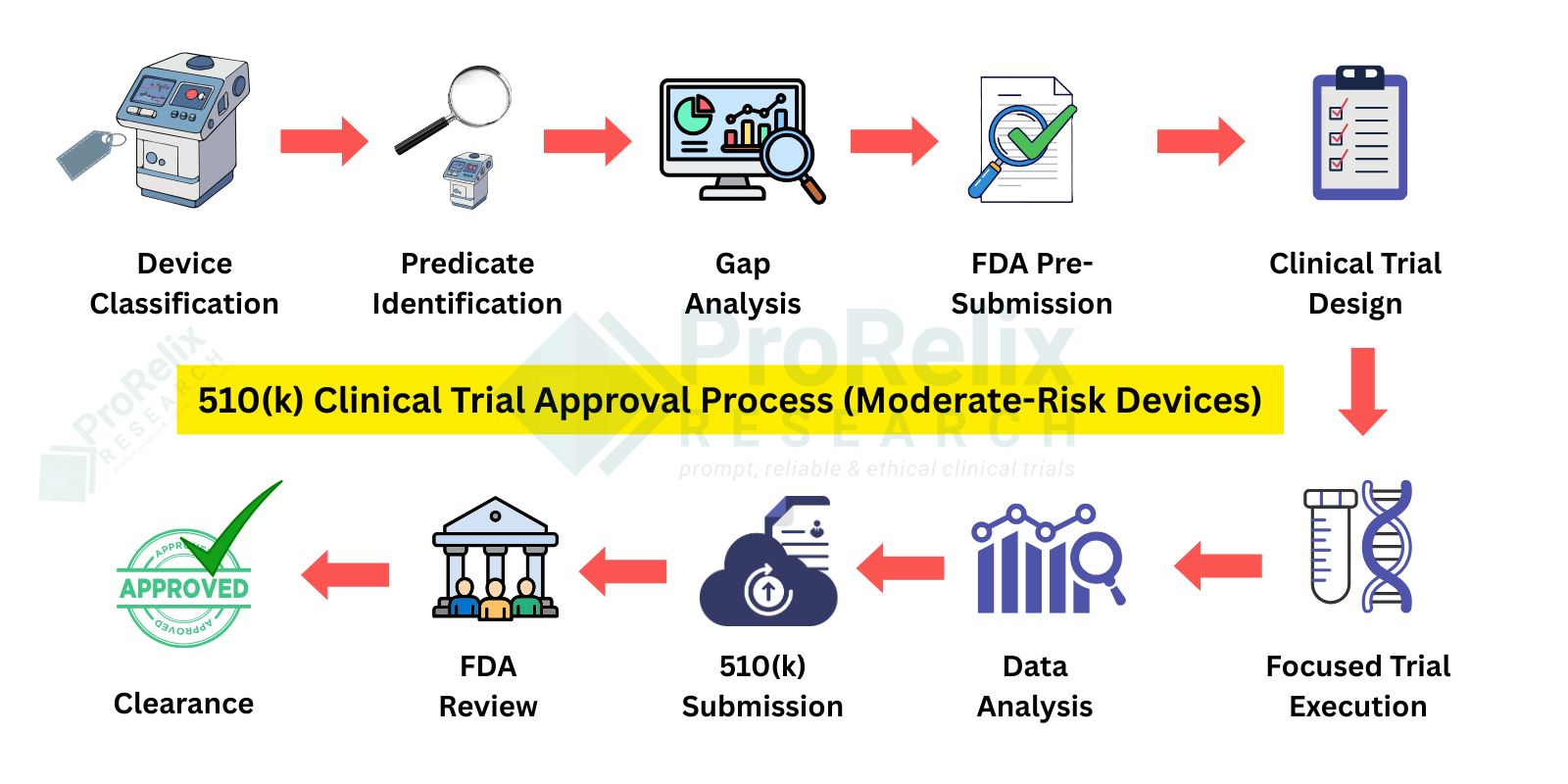
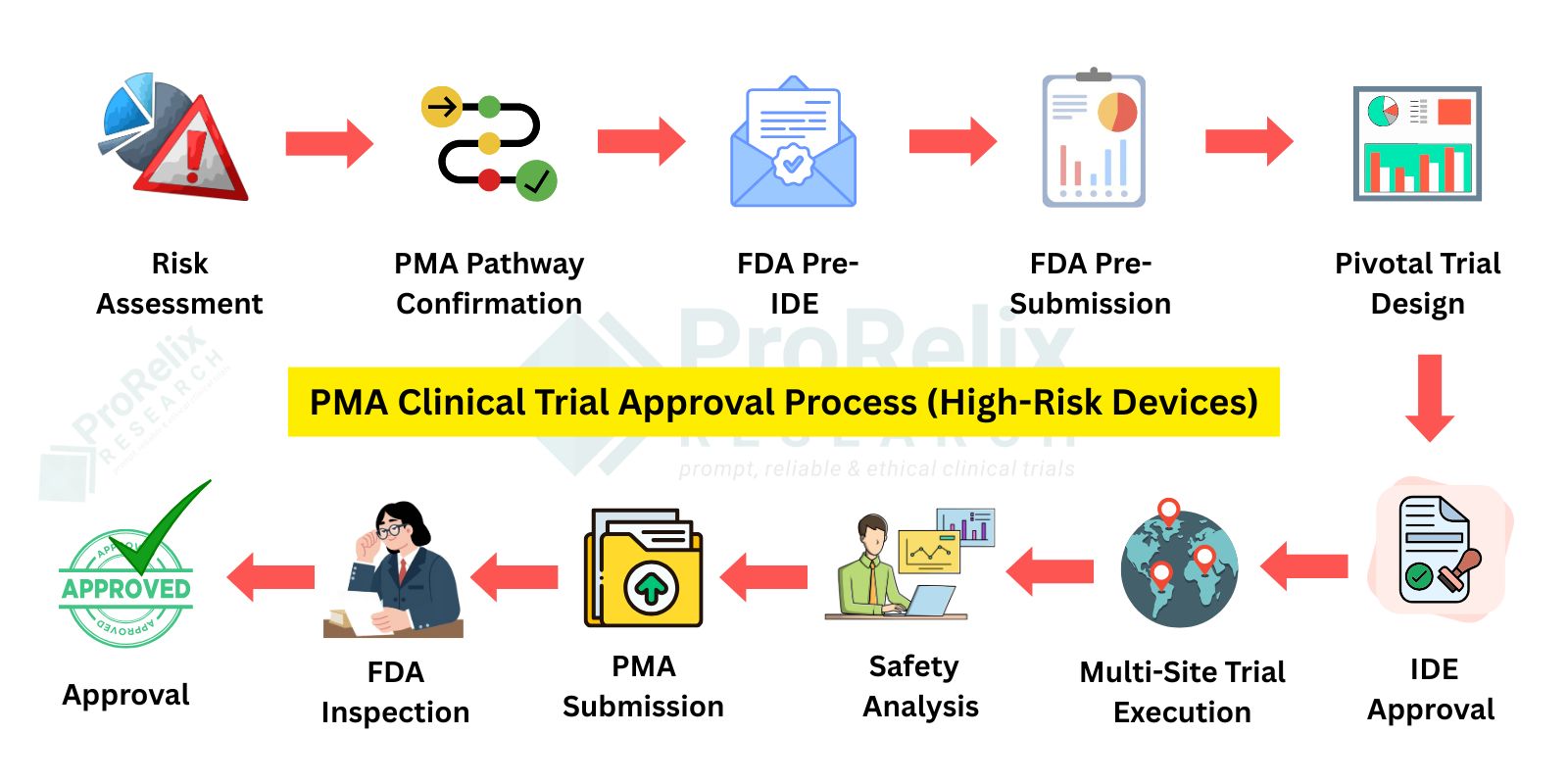
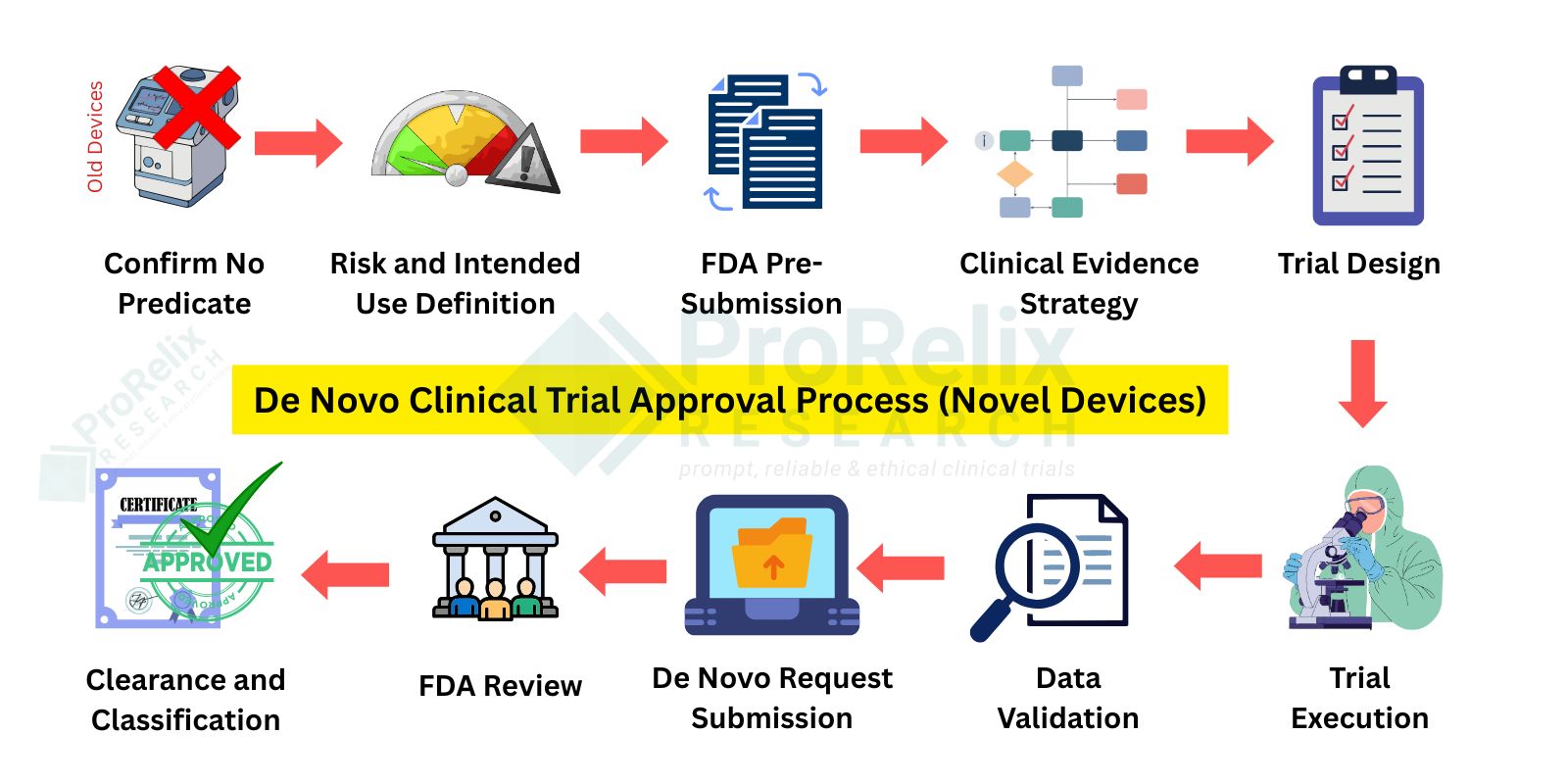
Navigating the regulatory pathways for medical devices requires precision, expertise, and a thorough understanding of FDA requirements. 510(k), PMA, and De Novo clinical trials each serve unique purposes: 510(k) trials demonstrate substantial equivalence to existing devices, PMA trials generate pivotal data for high-risk or implantable devices, and De Novo trials establish a new device classification when no predicate exists. Conducting these trials involves careful planning of protocols, site selection, patient recruitment, clinical monitoring, data collection, and analysis to ensure compliance with FDA guidelines, safety standards, and regulatory expectations. Comprehensive clinical evidence is critical to achieving FDA approval or clearance efficiently and successfully.
With years of global experience and a proven track record, Prorelix Research a reliable CRO provides 510 k, PMA and De Novo clinical trials services in the USA, India, and Europe with global regulatory compliance, submission-ready clinical evidence, and support to help medical device innovators accelerate approvals and bring safe, effective products to market.

Aspect | 510(k) Clinical Trial | PMA Clinical Trial | De Novo Clinical Trial |
Regulatory Purpose | Demonstrates substantial equivalence to a legally marketed predicate device | Demonstrates safety and effectiveness for high-risk devices | Establishes a new device classification for novel devices |
Device Risk Level | Moderate risk (Class II) | High risk (Class III) | Low to moderate risk (Class I or II) |
Predicate Device Required | Yes | No | No |
Clinical Trial Requirement | Sometimes required, depending on data gaps | Almost always required | Often required to support safety and performance |
Level of Clinical Evidence | Limited to moderate | Extensive and comprehensive | Moderate, focused on risk and performance |
Study Type | Comparative or performance-based studies | Pivotal, controlled clinical trials | Feasibility or pivotal studies |
IDE Requirement | Required if the study is a significant risk | Required | Required if significant risk |
FDA Review Time | Typically shorter | Longest review timeline | Moderate review timeline |
Regulatory Outcome | FDA Clearance | FDA Approval | FDA Clearance with new classification |
Submission Complexity | Moderate | Very high | High |
Cost and Resources | Lower compared to PMA | Highest cost and resource-intensive | Moderate cost |
Post-Market Requirements | Standard post-market controls | Often includes post-approval studies | Standard post-market controls |
Typical Applicants | Manufacturers with predicate devices | Manufacturers of life-sustaining or implantable devices | Innovators with first-of-its-kind devices |
Market Access Speed | Faster | Slowest | Moderate |
Future Predicate Use | Uses existing predicate | Does not create a predicate | Creates a new predicate for future devices |
The De Novo classification pathway was created to provide a regulatory route for novel, low-to-moderate risk medical devices that do not have a legally marketed predicate, allowing them to reach the market without requiring full PMA approval. During medical device clinical trials, the evaluation of unanticipated adverse device effects (UADEs) must be reported to the FDA by the sponsor or responsible party to ensure patient safety and regulatory compliance. Understanding the difference between FDA 510(k) clearance vs approval is also critical, as 510(k) clearance confirms substantial equivalence to an existing device, while FDA approval applies to PMA devices that require extensive clinical evidence to demonstrate safety and effectiveness.
Medical device sponsors must select the correct FDA pathway early in development to avoid delays, additional studies, or regulatory rejection. Choosing between 510(k), PMA, or De Novo depends on device risk, intended use, and available predicate devices, while strict reporting of unanticipated adverse device effects ensures ongoing patient safety. Proper regulatory planning, timely FDA communication, and compliance with reporting and documentation requirements play a critical role in achieving successful clearance or approval and maintaining long-term market compliance.

FDA 510(k) clearance requires medical device manufacturers to demonstrate that their device is substantially equivalent to a legally marketed predicate device in terms of intended use, technological characteristics, and performance. This process typically involves device classification, predicate identification, risk analysis, and the submission of scientific evidence such as bench testing, biocompatibility data, software validation, and, when necessary, clinical trial data. All documentation must align with FDA regulations, quality system requirements, and applicable guidance to support safety and effectiveness.
Meeting FDA 510(k) clearance requirements also includes preparing a complete and well-structured submission, responding to FDA questions during review, and ensuring post-clearance compliance. A strong regulatory and clinical strategy helps minimize review timelines, reduce deficiencies, and achieve faster market access for medical devices in the United States. Building on these regulatory and clinical requirements, ProRelix Research supports medical device innovators in meeting FDA 510(k) Clearance Requirements through expert clinical execution, regulatory strategy, and submission-ready evidence generation.
The 510(k), PMA, and De Novo approval processes require precise alignment with FDA regulatory expectations, clinical evidence standards, and risk-based decision making. Each pathway demands a structured approach covering trial design, data integrity, and regulatory documentation to achieve successful clearance or approval. ProRelix Research is a high-performing CRO for 510(k), PMA & De Novo Approval Process, supporting medical device innovators with advanced clinical execution and regulatory expertise across global markets.
Protocols define study objectives, endpoints, sample size, and methodology. Design must align with FDA guidance and device-specific risks. Well-designed trials ensure reliable and acceptable clinical outcomes.
Successful 510(k), PMA, and De Novo clinical trials require strict adherence to FDA guidelines, compliance standards, and regulatory certification requirements. Sponsors and CROs must ensure proper documentation, oversight, and inspection readiness to achieve regulatory approval or clearance efficiently and safely.
Adhering to FDA guidelines is essential for a successful clinical trial submission. A CRO for 510(k), PMA, De Novo guidelines ensures that study protocols, device classifications, and clinical documentation meet regulatory expectations. Following these structured guidelines reduces review timelines and supports smooth regulatory interactions.
Maintaining regulatory compliance throughout the trial lifecycle is critical for safety, data integrity, and FDA inspections. Working with a CRO for 510(k), PMA, De Novo compliance ensures continuous oversight of study conduct, documentation, and safety reporting, keeping the trial fully aligned with FDA requirements.
Preparing for FDA inspections and regulatory certification requires accurate documentation and robust quality systems. A CRO for 510(k), PMA, De Novo certification helps sponsors achieve inspection readiness, maintain audit trails, and comply with all applicable standards, increasing the likelihood of approval or clearance.
Understanding the regulatory landscape is critical for clinical trial success. A CRO for 510(k), PMA, De Novo regulations ensures that all FDA requirements are followed, from protocol design to reporting obligations, while minimizing regulatory risks and maintaining compliance throughout the trial.
The Transfer of Regulatory Obligations (TORO) ensures that sponsor responsibilities are formally reassigned during clinical trials, often from a sponsor to a CRO, while maintaining full regulatory compliance and accountability. Proper TORO management supports inspection readiness, continuous oversight, and regulatory transparency throughout the study lifecycle. ProRelix Research provides Transfer of Regulatory Obligations (TORO) services to supporting sponsors with accurate documentation, FDA-aligned processes, and compliant responsibility transitions across global clinical trials.
The FDA TORO form is the official document used to transfer regulatory responsibilities between a sponsor and a CRO. It ensures that all obligations, including trial oversight, safety reporting, and regulatory communications, are clearly assigned and recognized by the FDA. Completing this form accurately is critical to avoid regulatory gaps and ensure smooth operational continuity throughout the clinical trial. CROs specializing in 510(k), PMA, and De Novo trials often assist sponsors in preparing and submitting the TORO form efficiently.
Following transfer of regulatory obligations FDA guidance is essential to maintain regulatory compliance when responsibilities are reassigned. The guidance outlines when a TORO is necessary, how to document changes, and the roles and obligations of the new responsible party. Sponsors and CROs can leverage this guidance to mitigate compliance risks, ensure continuous oversight, and guarantee that trial objectives are not disrupted by organizational or operational changes.
TORO clinical trials require rigorous oversight to ensure all transferred responsibilities are executed according to FDA regulations. CROs provide structured monitoring, documentation, and compliance management to support sponsor accountability. Effective TORO oversight safeguards trial integrity, maintains inspection readiness, and ensures that clinical studies meet all regulatory standards for 510(k), PMA, and De Novo trials.
The FDA transfer of regulatory obligations form serves as the formal record for reassignment of sponsor responsibilities in clinical trials. It clearly documents accountability for trial management, regulatory reporting, safety oversight, and compliance activities. Accurate and timely completion ensures FDA acknowledgment of responsibility changes and supports transparent regulatory operations for sponsors and CROs, which is specialy critical for high risk or complex 510(k), PMA, and De Novo clinical trials conducted under FDA oversight.!
An FDA sponsor inspection checklist helps sponsors and CROs prepare for regulatory audits and ensure all responsibilities are documented and compliant. The checklist typically covers trial oversight, data integrity, safety reporting, and documentation management. Using this tool, sponsors can proactively identify gaps, maintain FDA compliance, and ensure that transferred responsibilities under TORO are effectively managed throughout the study lifecycle.
The FDA guidance for clinical trial sponsors outlines expectations for trial oversight, data monitoring, safety reporting, and regulatory compliance. Following this guidance ensures that both the sponsor and the CRO maintain alignment on responsibilities, particularly when obligations are transferred. This structured approach improves trial quality, facilitates timely regulatory approvals, and reduces inspection findings.
Regulatory affairs services help pharmaceutical and medical device companies comply with national and international regulations to gain product approval. These services include documentation, submission management, and communication with health authorities such as the FDA, EMA, and CDSCO.
Regulatory affairs consulting ensures that all clinical trial activities meet global regulatory standards. At ProRelix Research, our experts assist with Clinical Trial Applications (CTA), Investigational New Drug (IND) submissions, and ongoing compliance to ensure trials start on time and run smoothly.
A PMA clinical trial is a comprehensive, FDA-regulated study required for high-risk Class III medical devices to demonstrate safety and effectiveness. A CRO supports PMA trials by managing complex study designs, regulatory submissions, monitoring, data integrity, and inspection readiness to meet stringent FDA requirements.
A De Novo clinical trial is conducted for novel medical devices with no existing predicate but classified as low to moderate risk. CRO involvement is important to generate clinical evidence, support FDA classification, establish special controls, and ensure compliance throughout the De Novo approval process.
A CRO ensures that clinical trials comply with FDA regulations, guidance documents, and submission requirements. This includes protocol alignment, regulatory documentation, safety reporting, monitoring, and maintaining inspection-ready systems across all FDA pathways.
A CRO provides end-to-end services including clinical trial planning, site selection, monitoring, data management, regulatory support, quality assurance, and FDA inspection preparation for 510(k), PMA, and De Novo clinical trials.
A sponsor should engage a CRO when clinical data is required for FDA clearance or approval, especially when internal regulatory or clinical expertise is limited. Early CRO involvement helps reduce delays, manage regulatory risk, and improve trial execution efficiency.
A CRO supports FDA inspections by maintaining compliant documentation, implementing quality systems, conducting internal audits, and ensuring adherence to FDA guidance. This helps sponsors remain inspection-ready throughout 510(k), PMA, and De Novo clinical trials.
A 510(k) clinical trial is done to show that a medical device works in a similar way to an already approved device, and it usually needs less clinical data. A PMA clinical trial is required for high-risk medical devices and involves detailed clinical studies to prove the device is safe and effective before FDA approval. A De Novo clinical trial is used for new, low-to-moderate risk devices that do not have an existing comparison device and helps the FDA decide how the device should be classified and cleared.
The term 510(k) refers to Section 510(k) of the U.S. FDA Act, which is a regulatory pathway and not a monetary value. In terms of cost, a 510(k) submission typically ranges from USD 12,000 to 25,000 in FDA fees, which is approximately ₹10 to ₹21 lakhs (INR), excluding clinical trial, testing, and CRO service costs. The total overall expense may vary depending on device complexity, clinical data requirements, and regulatory support needed.
Bringing new therapies requires accuracy, efficiency, and regulatory care. We offer clinical trial solutions that reduce risk, shorten timelines, and ensure compliance, turning ideas into successful outcomes.

Be the first to know the latest trends in clinical research, real-world case studies, and industry secrets.

ProRelix Research is the rapidly growing Contract/ Clinical Research Organization (CRO) with multi-country service capability supporting phase 1, 2, 3, & 4 clinical trials of Pharma, Biotech, Biopharma, Medical Device, Nutraceutical & Herbal companies to conduct in the USA, India, Europe & South East Asia.