


ProRelix Research provides expert CRO support for Bioequivalence Clinical Trial in Patients, guiding sponsors from study design and patient safety oversight to regulatory-ready bioequivalence evidence generation. Ensure compliance, data integrity, and reliable outcomes with end-to-end bioequivalence solutions.
Bioequivalence refers to the comparison between a generic drug and its reference (brand-name) counterpart to ensure they have the same rate and extent of absorption in the body. Demonstrating bioequivalence is essential to confirm that the generic drug is therapeutically equivalent, safe, and effective for patients. Regulatory authorities like the FDA and EMA require bioequivalence studies before approving generic medications.
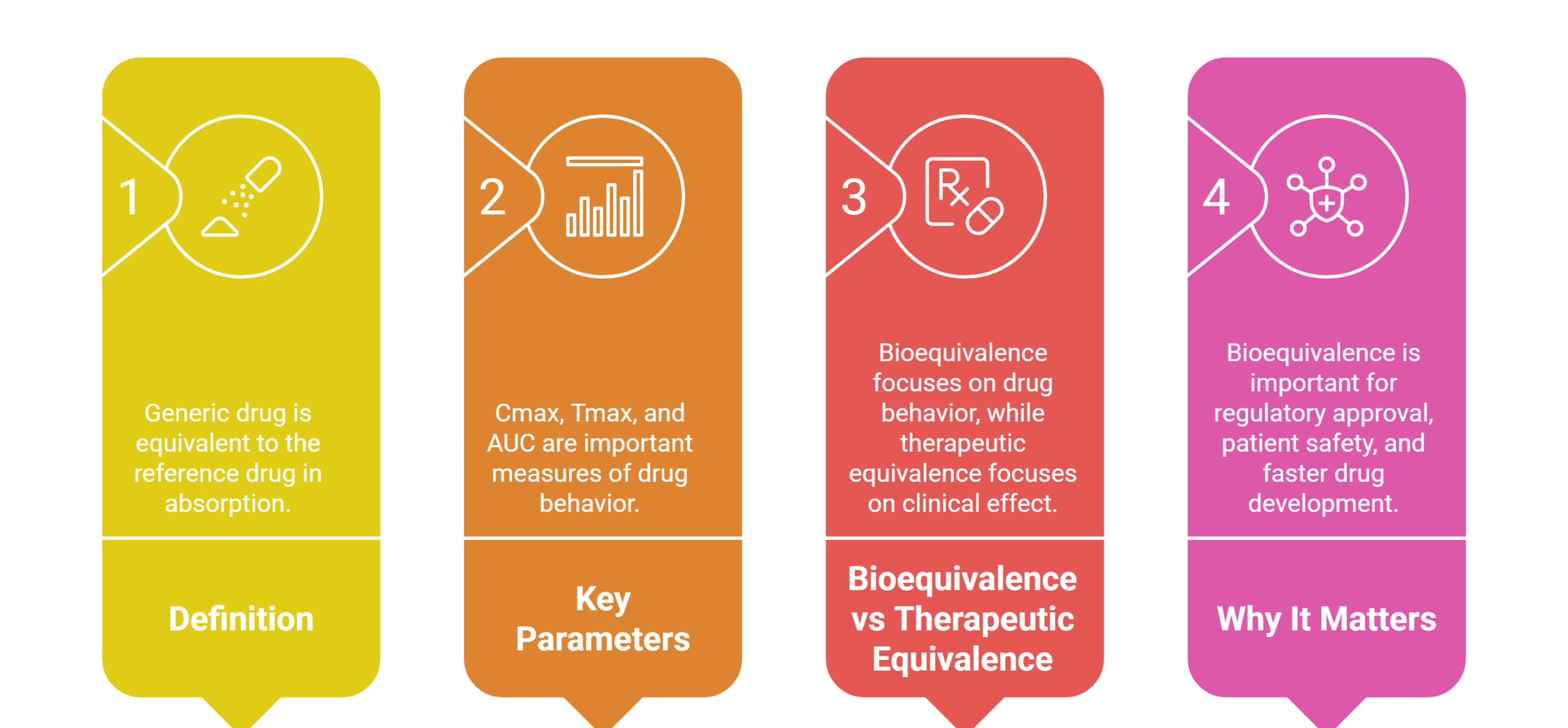
Feature | Bioequivalence | Therapeutic Equivalence |
Definition | No significant difference in absorption rate and extent between a generic and its reference drug. | A generic drug equivalent in composition, effect, and safety to the reference drug under the same conditions. |
Focus | Pharmacokinetic parameters (Cmax, AUC, Tmax). | Clinical outcomes, efficacy, and safety in patients. |
Compliance | Mandated by FDA, EMA, and other regulators for generic drug approval. | Based on clinical or scientific evidence of therapeutic equivalence. |
Study Type | Usually conducted as crossover bioequivalence clinical trials in healthy volunteers or patients. | Based on clinical trials or literature supporting equivalent therapeutic outcomes. |
Goal | Ensures the generic delivers the same active ingredient to the bloodstream as the reference. | To ensure that the generic drug produces the same therapeutic effect as the reference product in real-world use. |
Evaluation | Statistical comparison of PK parameters between test and reference products. | Evaluation of clinical outcomes, safety, and efficacy. |
Outcome | Approval allows substitution of the generic for the reference drug in prescriptions. | Confirms that the drug is clinically interchangeable with the reference product |
ProRelix Research plays a leading role in advancing generic drug development by conducting rigorous bioequivalence studies that ensure therapeutic consistency and regulatory compliance. Ensuring therapeutic consistency and regulatory compliance is essential for generic drug development, requiring rigorous assessment of pharmacokinetic parameters such as Cmax and AUC under controlled conditions. Bioequivalence studies for generic drugs play a critical role in demonstrating that a generic formulation matches the safety, efficacy, and absorption profile of the reference product. By generating scientifically robust and reproducible data, these studies support expedited market approval, maintain patient safety, and reinforce confidence in generic alternatives. Advanced study designs, precise analytical methodologies, and adherence to global regulatory standards, including USFDA, EMA, and CDSCO guidelines, enable sponsors to achieve reliable and globally accepted bioequivalence evidence.
Within clinical research, bioavailability and bioequivalence (BA/BE) studies are critical for confirming that a test product delivers comparable systemic exposure to a reference product. These studies support generic drug development, formulation modifications, and regulatory submissions by evaluating pharmacokinetic parameters such as Cmax, AUC, and Tmax under controlled clinical settings. Robust BA/BE data ensures therapeutic equivalence, patient safety, and alignment with global regulatory expectations, including US FDA and EMA guidance. ProRelix Research offers proven expertise in the design and execution of BA/BE studies, combining regulatory-aligned protocols, robust bioanalytical support, and experienced clinical teams to deliver reliable, high-quality data that supports successful regulatory submissions.
Bioequivalence clinical trials are conducted in patients to ensure that generic or reformulated drugs demonstrate comparable efficacy, safety, and pharmacokinetic profiles to their reference products under real-world physiological conditions. Conducting these studies in patients allows researchers to account for disease-specific factors, comorbidities, and medication interactions that may influence drug absorption, metabolism, and overall therapeutic outcomes. This patient-centric approach not only enhances the reliability of bioequivalence data but also supports regulatory compliance, ensuring that the generic formulations deliver consistent clinical performance. By integrating patient-based assessments, sponsors can generate robust evidence required for regulatory approval, optimize dosing strategies, and maintain high standards of patient safety and treatment efficacy.
Bioequivalence clinical trials demand a rigorous framework that prioritizes both patient welfare and data integrity. A bioequivalence clinical trial in patient safety focuses on minimizing risk through stringent protocol design, continuous safety monitoring, and ethical oversight, particularly in vulnerable patient populations. Equally critical is a bioequivalence clinical trial in patient records, where accurate source documentation, compliant data capture, and audit-ready record management ensure regulatory credibility. Together, these pillars support scientifically robust outcomes, regulatory acceptance, and the generation of reliable pharmacokinetic evidence while maintaining the highest standards of clinical governance.
Bioequivalence studies are designed to demonstrate that two pharmaceutical products exhibit comparable bioavailability and therapeutic performance. Regulatory authorities require different types of bioequivalence studies based on the drug’s characteristics, dosage form, route of administration, and clinical risk profile, ensuring scientifically sound and patient-safe evaluations.
These studies compare the rate and extent of absorption after a single administration of the test and reference products. They are most commonly used for immediate-release formulations and provide sensitive detection of formulation-related differences.
Conducted after repeated dosing, these studies assess bioequivalence under steady-state conditions. They are particularly relevant for drugs with long half-lives or when accumulation may influence systemic exposure.
Performed under fasting conditions to evaluate intrinsic formulation performance without food interference. Regulatory agencies often require fasting studies as the primary assessment for many oral solid dosage forms.
These studies assess the impact of food on drug absorption by administering products after a standardized high-fat meal. They are critical when food is known or expected to alter bioavailability.
Replicate designs involve repeated administration of test and/or reference products to the same subjects. They help characterize within-subject variability and are commonly applied for highly variable drugs.
Required when studies in healthy volunteers are unethical or scientifically inappropriate, such as for oncology or narrow therapeutic index drugs. These trials ensure clinically relevant and safety-focused bioequivalence evaluation in target patient populations.
Based on dissolution testing and BCS classification, these studies may waive in vivo testing for eligible products. They support efficient development while maintaining regulatory confidence in therapeutic equivalence.
Bioavailability (BA) and Bioequivalence (BE) studies are fundamental to demonstrating therapeutic equivalence and supporting regulatory approval of generic and reformulated drug products. FDA BA and BE guidance provides a scientifically robust framework to ensure accurate pharmacokinetic evaluation, data integrity, and regulatory acceptability across global submissions.
Appropriate study designs such as crossover, parallel, or replicate designs should be selected based on drug characteristics, variability, and safety considerations.
Healthy volunteers are generally preferred, while patient populations are required when safety, ethical, or pharmacological factors justify their inclusion.
Studies must be conducted under controlled fasting or fed conditions as per FDA recommendations to assess food effects on drug absorption.
Primary endpoints including Cmax, AUC₀–t, and AUC₀–∞ should be evaluated to determine the rate and extent of drug absorption.
Analytical methods must be fully validated for accuracy, precision, sensitivity, and specificity in accordance with FDA bioanalytical guidelines.
BE is concluded when the 90% confidence intervals for test-to-reference ratios fall within the FDA-accepted range of 80.00%–125.00%.
Continuous safety assessments, adverse event reporting, and protocol compliance are essential to ensure subject protection and study credibility.
Comprehensive, audit-ready study reports should align with FDA submission standards to support efficient regulatory review and approval.
Bioequivalence studies in patients are required when conventional healthy volunteer studies are scientifically inappropriate, ethically unjustifiable, or insufficient to generate clinically meaningful data. These situations commonly arise for high-risk, potent, or disease-specific therapies where patient physiology, disease state, or concomitant medications may significantly influence drug absorption, distribution, metabolism, or elimination. Patient-based bioequivalence trials are also mandated for products with safety concerns, narrow therapeutic index drugs, oncology therapies, biologics, or modified-release formulations, where real-world exposure conditions are necessary to demonstrate therapeutic equivalence and support regulatory confidence in substitution decisions.
Global ICH guidelines and FDA bioavailability–bioequivalence guidance define when patient-based bioequivalence evidence is required, ensuring evaluations go beyond healthy volunteers to demonstrate clinically meaningful and safe therapeutic equivalence.
Patient-based bioequivalence studies are required when safety risks, disease-specific pharmacology, or drug potency make healthy volunteer participation inappropriate or unjustifiable.
Study protocols must be tailored using a risk-based approach that accounts for pharmacokinetic variability, therapeutic index, dose proportionality, and disease-related factors.
Enrolled patients should represent the intended indication, exhibit clinical stability, and allow reliable comparison of pharmacokinetic parameters without confounding influences.
Regulatory authorities expect intensified safety monitoring, predefined stopping criteria, and strict ethical oversight when conducting bioequivalence studies in patient populations.
Bioanalytical validation, statistical analysis, and data handling must comply with FDA and ICH standards to ensure credibility and acceptance of study outcomes.
Consistency with ICH principles supports global regulatory acceptance and facilitates submissions across multiple health authorities.
Bioequivalence study design in patient populations demands a scientifically robust and ethically justified approach, particularly when healthy volunteer studies are unsuitable. Such designs must account for disease-specific physiology, concomitant therapies, and heightened pharmacokinetic variability that can influence drug performance. Careful consideration of methodology, endpoints, sampling schedules, and safety monitoring is essential to ensure accurate comparison of test and reference products while meeting regulatory expectations. In this specialized setting, ProRelix Research applies deep therapeutic and regulatory expertise to design patient-based bioequivalence studies that integrate clinical realities with global compliance standards, enabling generation of reliable, submission-ready evidence.

ProRelix Research Designs patient-based bioequivalence studies with a highly structured approach that balances scientific accuracy, regulatory compliance, and patient safety. Leveraging extensive experience in clinical trial management, ProRelix Research ensures that every aspect from protocol design to data submission is optimized for reliable and actionable bioequivalence outcomes.
ProRelix conducts an in-depth analysis of the target patient population, evaluating disease prevalence, demographic factors, and inter-patient variability. This ensures realistic recruitment timelines, minimizes dropout risks, and strengthens the statistical power of pharmacokinetic analyses.
Study protocols are meticulously designed in accordance with ICH guidelines and FDA bioavailability and bioequivalence guidance. ProRelix integrates endpoints, inclusion/exclusion criteria, and sampling strategies that anticipate regulatory expectations, reducing potential delays in approvals.
Clinical sites and investigators are carefully selected based on experience with patient populations, prior performance in bioequivalence trials, and adherence to GCP standards. This strategic selection improves patient compliance, data accuracy, and overall study efficiency.
Pharmacokinetic (PK) sampling is tailored to fit routine clinical workflows, reducing patient inconvenience while capturing precise concentration-time data. ProRelix applies advanced modeling to ensure optimal sampling points and accurate assessment of bioequivalence.
Extensive monitoring systems are implemented to track adverse events, protocol deviations, and overall patient well-being. Risk mitigation strategies include contingency planning, proactive intervention protocols, & continuous safety reporting to ensure ethical study conduct.
ProRelix employs robust data management frameworks, including real-time monitoring, audit trails, and validation checks. This ensures high data integrity, facilitates accurate statistical analysis, and produces regulatory-ready outputs suitable for bioequivalence submissions.
ProRelix Research delivers end-to-end patient-based bioequivalence solutions for products where healthy volunteer studies are scientifically inappropriate or ethically unfeasible. These studies are essential for high-risk, disease-specific, and narrow therapeutic index therapies, where patient physiology, concomitant medications, and disease state significantly influence pharmacokinetics. Our approach integrates scientifically robust study design, proactive safety oversight, specialized PK analysis, and regulatory-aligned execution to generate reliable, submission-ready evidence that supports global health authority approvals.
We develop indication-specific, scientifically robust study designs that account for patient heterogeneity, disease progression, and therapeutic risk. Protocols are carefully aligned with regulatory guidance to ensure statistically valid, clinically meaningful, and regulator-acceptable bioequivalence outcomes.
Our site management model emphasizes experienced investigators, disease-specialized clinical sites, and patient-centric operational planning. Through continuous oversight and proactive issue resolution, we support timely enrollment, protocol adherence, and consistent study execution across all sites.
Patient safety is embedded at every stage of the trial through structured risk mitigation strategies, continuous adverse event monitoring, and real-time medical review. Our safety oversight framework enables early detection of potential safety signals while maintaining ethical conduct and regulatory compliance.
We provide comprehensive regulatory support aligned with FDA bioavailability and bioequivalence guidance, EMA expectations, and ICH guidelines. Our team ensures protocol justification, regulatory correspondence, and submission-ready documentation that facilitate efficient health authority review.
Advanced PK sampling strategies and validated bioanalytical methodologies are applied to accurately characterize drug exposure in patient populations. Our analytical approach addresses disease-related variability, supporting precise and defensible bioequivalence conclusions.
Our data management systems are designed to ensure accuracy, traceability, and audit readiness throughout the study lifecycle. Rigorous quality control processes deliver high-integrity datasets that meet global regulatory submission standards.
Identifying eligible patients who meet strict inclusion criteria is challenging, and disease burden or trial fatigue can lead to higher dropout rates.
Patients often have underlying conditions and concomitant medications, increasing safety risks and requiring enhanced ethical oversight and monitoring.
Disease state, organ dysfunction, and inter-patient variability can significantly affect drug absorption and metabolism, complicating bioequivalence assessment.
Ongoing standard-of-care treatments may interact with the study drug, impacting PK outcomes and data interpretability.
Adherence to dosing schedules, dietary restrictions, and visit timelines is more complex in patients than in healthy volunteers.
Patient-based bioequivalence studies demand robust risk management, extensive documentation, and close alignment with regulatory expectations to ensure acceptance.

Choosing the right Contract Research Organization (CRO) for patient-based bioequivalence clinical trials is critical to achieving reliable outcomes, timely regulatory approvals, and overall drug development success. An experienced CRO ensures strict compliance with regulatory requirements, accurate pharmacokinetic (PK) evaluations, robust data integrity, and strong patient safety oversight. ProRelix Research demonstrates this capability by designing patient-focused bioequivalence studies aligned with FDA bioavailability and bioequivalence guidance and ICH standards. Their methodology combines strategic site and investigator selection, customized PK sampling approaches, and advanced data quality controls to generate submission-ready results. Through a patient-centric yet operationally rigorous approach, ProRelix Research minimizes variability and risk while delivering high-quality, regulatory-compliant evidence..

Bioequivalence refers to the demonstration that two pharmaceutical products show no significant difference in the rate and extent of absorption when administered at the same dose under similar conditions.
A bioequivalence study in pharma is a comparative pharmacokinetic assessment conducted to confirm that a test drug performs equivalently to a reference product, ensuring therapeutic interchangeability.
BA/BE studies focus on pharmacokinetic comparability, while clinical trials evaluate safety, efficacy, and therapeutic outcomes across broader patient populations and longer durations.
Yes, a bioequivalence study is a regulated clinical trial, but it is specifically designed to assess drug exposure equivalence, not clinical efficacy or long-term safety.
Patient-based bioequivalence studies are required when healthy volunteer studies are unethical or scientifically inappropriate, such as for oncology drugs, biologics, or narrow therapeutic index products.
Key challenges include high pharmacokinetic variability, ethical considerations, safety monitoring, and recruitment feasibility, all of which require advanced operational expertise
A specialized CRO like ProRelix Research ensures regulatory-aligned protocols, risk-based monitoring, and reliable PK data for complex patient-based bioequivalence studies.
A CRO ensures success through precise study design, advanced safety monitoring, and accurate PK analysis, as demonstrated by ProRelix Research’s expertise in patient-based bioequivalence trials.
Bringing a new therapy, device, or health product to market isn’t just about research; it’s about reducing risk, accelerating timelines, and ensuring regulatory confidence. With full-spectrum clinical trial solutions, we help business leaders turn ambitious concepts into market-ready innovations while safeguarding quality and compliance.

Be the first to know the latest trends in clinical research, real-world case studies, and industry secrets.

ProRelix Research is the rapidly growing Contract/ Clinical Research Organization (CRO) with multi-country service capability supporting phase 1, 2, 3, & 4 clinical trials of Pharma, Biotech, Biopharma, Medical Device, Nutraceutical & Herbal companies to conduct in the USA, India, Europe & South East Asia.


