


Support your biological and biosimilar clinical trials with our experienced CRO services, from protocol development and immunogenicity assessment to global regulatory submissions. Our integrated approach ensures compliance, efficiency, and high-quality clinical evidence.
Biologics and biosimilars clinical studies designs due to their complex structures, sensitive manufacturing processes, and stringent regulatory expectations. We bring deep scientific expertise and global regulatory knowledge to design and manage biologic and biosimilar clinical trials that meet the highest standards of safety, efficacy, and compliance.
ProRelix Research is a trusted and high performing CRO for biological and biosimilar clinical trials in USA, India and Europe to support sponsors across all phases of development from early phase studies to pivotal and post-marketing trials. Our team designs scientifically robust protocols aligned with FDA, EMA, and ICH guidelines, ensuring comparability, immunogenicity assessment, and data integrity throughout the trial lifecycle.
Our end-to-end biologics and biosimilar clinical trial design services include protocol development, regulatory strategy, site feasibility, patient recruitment, clinical monitoring, pharmacovigilance, and data management. By combining scientific precision with operational excellence. We helps sponsors generate high-quality clinical evidence and successfully advance biologics and biosimilars to market with confidence.
Biological and biosimilar clinical products play a important role in modern healthcare, offering targeted therapies for complex and chronic diseases. Due to their complexity and sensitivity, clinical trials involving biologics and biosimilars must follow stringent regulatory, scientific, and ethical standards. A CRO for Biological and Biosimilar Clinical Trials Guidelines ensures that these clinical studies are designed and conducted in full compliance with global regulatory expectations, safeguarding patient safety, data integrity, and product quality. These guidelines govern every stage of development from preclinical studies and clinical trial execution to regulatory submissions and post-marketing surveillance.
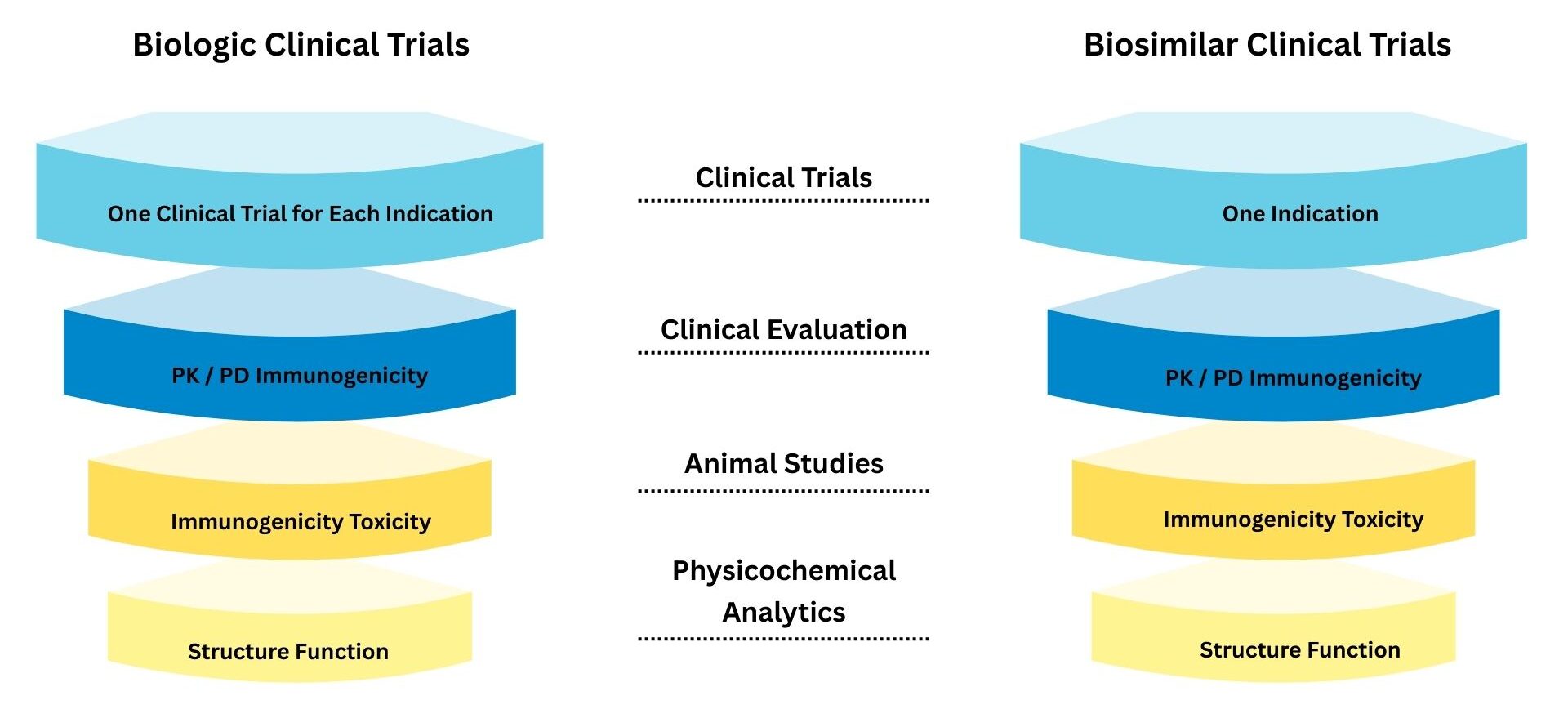
Clinical trials for biological and biosimilar products must comply with global and local regulatory guidelines such as ICH-GCP, WHO, EMA, FDA, and national authorities to ensure safety, quality, and efficacy.
Biosimilar trials are designed to demonstrate high similarity to the reference biologic through stepwise analytical, non-clinical, and clinical evaluations, focusing on minimizing clinically meaningful differences.
Trials typically follow a phased approach (Phase I–III), emphasizing pharmacokinetics (PK), pharmacodynamics (PD), immunogenicity, and confirmatory efficacy studies when required.
Well-defined study designs, sensitive patient populations, and clinically relevant endpoints are selected to effectively detect differences between the biosimilar and reference product.
Monitoring anti-drug antibodies (ADA) is critical, as immunogenicity can impact safety, efficacy, and long-term clinical outcomes of biological and biosimilar products.
Robust safety monitoring, pharmacovigilance plans, and risk mitigation strategies are implemented throughout the trial lifecycle to protect patient well-being.
Consistent manufacturing processes, validated analytical methods, and strict quality controls are essential to ensure product stability and batch-to-batch consistency during clinical trials.
Ethical approval, informed consent, and patient rights protection are mandatory, with special attention to vulnerable populations and long-term exposure risks.
Accurate data collection, validation, and regulatory-ready documentation are required to support trial outcomes and submission to health authorities.
Post-marketing clinical studies and ongoing safety surveillance may be required to confirm long-term safety, efficacy, and interchangeability where applicable.
Biologicals and biosimilars follow structured clinical trial phases to demonstrate safety, efficacy, quality, and comparability. While the overall phases are similar, biosimilar trials focus more on proving similarity to the reference product rather than re-establishing full efficacy from scratch.

Biological and biosimilar products are among the most scientifically sophisticated therapeutic modalities in modern healthcare, developed using complex living systems rather than chemically synthesized compounds. Their large molecular structures, sensitivity to manufacturing processes, and inherent biological variability introduce unique challenges across clinical development. As a result, their evaluation demands a highly structured, evidence-driven approach supported by advanced analytical characterization, functional assessments, and carefully designed clinical studies. Biosimilar clinical drug development, in particular, is governed by a stepwise comparability paradigm that integrates analytical, non-clinical, and clinical data to establish similarity to a licensed reference product without unnecessary duplication of clinical exposure.
ProRelix Research supports Biological and Biosimilar Clinical Trial Requirements through structured clinical research services aligned with global regulatory standards. As a global CRO for Biological and Biosimilar Clinical Trials, its involvement includes analytical comparability planning, protocol development, immunogenicity assessment, clinical trial execution, regulatory documentation, and pharmacovigilance support. This approach helps ensure scientifically sound study design, regulatory compliance, and reliable data generation across development phases.
Clinical trials for biologics and biosimilars must comply with international and regional regulatory guidelines such as ICH, WHO, FDA, EMA, and local health authorities. Biosimilar development follows a stepwise approach emphasizing comparability rather than independent efficacy.
Extensive analytical studies are required to demonstrate structural, functional, and physicochemical similarity between the biosimilar and the reference biologic. These studies form the foundation before initiating clinical trials.
Preclinical studies assess pharmacodynamics, toxicity, and immunogenicity. For biosimilars, non-clinical requirements are often reduced if analytical similarity is well established.
Clinical development typically includes:
Phase I: Pharmacokinetic (PK) and pharmacodynamic (PD) comparability studies
Phase III: Confirmatory clinical studies to demonstrate equivalent efficacy, safety, and immunogenicity
Phase II trials may be waived for biosimilars depending on regulatory acceptance.
Immunogenicity evaluation is a critical requirement, involving the assessment of anti-drug antibodies (ADA) and neutralizing antibodies, as these can impact safety and therapeutic efficacy.
Equivalence or non-inferiority study designs are commonly used. Primary endpoints are sensitive clinical markers capable of detecting differences between the biosimilar and reference product.
Clinical trials must be conducted in sensitive patient populations and indications where differences, if any, are most likely to be detected. Extrapolation of indications may be allowed based on scientific justification.
A comprehensive post-marketing surveillance and risk management plan is mandatory to monitor long-term safety and rare adverse events after approval.
All clinical trials must strictly adhere to GCP standards, ensuring ethical conduct, patient safety, data integrity, and regulatory compliance throughout the study lifecycle.
Accurate clinical study reports, comparability data, and regulatory dossiers (CTD/eCTD) are essential for successful approval of biological and biosimilar products.
ProRelix Research provides approval process support for biological and biosimilar clinical trials, helping sponsors align study planning and documentation with regulatory requirements while meeting strict scientific, clinical, and ethical evaluation standards.
Before human clinical studies begin, sponsors must generate robust preclinical data to support product safety and scientific validity. This includes comparative analytical characterization, in vitro and in vivo toxicity studies, and pharmacokinetic and pharmacodynamic assessments to evaluate product behavior and biological activity. These studies form the foundation for obtaining clinical trial authorization. We supports sponsors in conducting these essential preclinical and analytical assessments in line with regulatory requirements.
A well-defined regulatory strategy is crucial for biosimilar development. Early engagement with regulatory agencies helps clarify study design expectations, comparability requirements, clinical data needs, & the scope for extrapolation of indications. ProRelix Research provides tailored regulatory guidance for biologics & biosimilars, supporting efficient development & approval across global markets.
Sponsors must submit a Clinical Trial Application (CTA) or Investigational New Drug (IND) dossier that includes Quality (CMC) documentation, non-clinical study reports, a clinical trial protocol, an investigator brochure, & a risk–benefit analysis. Regulatory authorities review these documents to ensure scientific validity & participant safety. We supports sponsors by assisting in the preparation & regulatory alignment of CTA/IND submissions to enable smooth & timely approvals.
Independent Ethics Committees (ECs) or Institutional Review Boards (IRBs) play a critical role in reviewing clinical trials to ensure the rights, safety, and well-being of study participants are fully protected. Their approval process emphasizes key areas such as the thoroughness of the informed consent process, the implementation of robust patient safety measures, and the ethical justification of the overall study design. We support in facilitating these reviews, ensuring that all trial protocols meet the highest ethical standards while prioritizing participant welfare.
Health authorities like the DCGI (India), FDA (US), EMA (EU), and other national agencies carefully review clinical trial applications for biological and biosimilar products, ensuring all regulatory, ethical, and safety requirements are met. ProRelix Research supports sponsors through this process, providing expert guidance to help secure timely approvals while maintaining compliance and participant safety.
After approval, clinical trials must follow Good Clinical Practice (GCP) guidelines, ensure proper safety reporting and pharmacovigilance, and implement protocol amendments and regulatory updates. ProRelix Research provides full support to maintain compliance throughout the trial lifecycle.
Biologics have transformed the management of complex conditions such as autoimmune diseases and cancer by providing targeted, effective therapies, though their high costs can limit patient access. Biosimilars highly similar versions of approved biologics offer the same therapeutic benefits at lower costs, making advanced treatments more accessible and sustainable within healthcare systems.
Integrating biosimilars into clinical practice requires careful consideration of safety, efficacy, interchangeability, and patient education. Their adoption influences prescribing patterns, treatment strategies, and healthcare economics, highlighting the importance of staying informed on emerging biosimilars and evolving clinical guidelines to ensure safe, effective, and cost-efficient patient care.
Biological & biosimilar products are complex medicines that require strict regulatory oversight during clinical development. Regulatory frameworks ensure that these trials are conducted safely, ethically, & with scientific rigor, protecting both participants & public health.
All trials are conducted in accordance with ICH-GCP (International Council for Harmonisation – Good Clinical Practice) standards, ensuring ethical conduct, participant safety, and generation of reliable, high-quality data.
Before initiating any study, necessary approvals are obtained from national regulatory authorities such as the FDA, EMA, or CDSCO, along with institutional ethics committees, ensuring the trial meets all legal and ethical requirements.
Continuous monitoring of adverse events and timely safety reporting are implemented throughout the trial to protect participants and ensure regulatory compliance.
Every trial is conducted strictly according to the approved protocol, and any modifications are documented, reviewed, and approved by regulatory authorities and ethics boards to maintain scientific integrity.
Biological and biosimilar products require careful handling, storage, and documentation. Strict quality management ensures product consistency and traceability throughout the trial lifecycle.
ProRelix Research provides end-to-end guidance and management for biological and biosimilar trials, helping sponsors navigate complex regulations efficiently while maintaining the highest standards of compliance, safety, and data integrity.

Biological and biosimilar drugs are advanced therapies derived from living organisms, requiring rigorous clinical trials to ensure safety, efficacy, and regulatory compliance. Careful study design, monitoring of adverse events, and strict data management maintain trial integrity and support timely approvals. Research on these drugs helps advance innovative treatments while safeguarding patient well-being and producing reliable scientific evidence.
Sr No | Features | Biologics Clinical Trials | Biosimilar Clinical Trials |
1 | Definition | Clinical trials for original biological drugs developed from living organisms. | Clinical trials for drugs highly similar to an already approved biologic, with no clinically meaningful differences in safety, purity, or potency. |
2 | Objective | To evaluate safety, efficacy, and optimal dosing of a new biologic. | To demonstrate similarity to the reference biologic in terms of efficacy, safety, and immunogenicity. |
3 | Preclinical Phase | Extensive preclinical studies are required to assess safety and mechanism of action. | Usually limited preclinical studies, focusing on comparative analysis with the reference product. |
4 | Clinical Phase I | Focuses on safety, tolerability, and pharmacokinetics in healthy volunteers or patients. | Often shorter; may involve fewer participants to compare pharmacokinetics (PK) and pharmacodynamics (PD) with the reference drug. |
5 | Clinical Phase II & III | Large-scale trials to determine efficacy, dosing, and side-effect profile in target patient population. | Trials focus on equivalence or non-inferiority to the reference biologic rather than establishing efficacy from scratch. |
| 6 | Regulatory Requirement | Full regulatory submission with complete data on safety, efficacy, and quality. | Abbreviated pathway; relies heavily on existing data from the reference biologic along with comparative studies. |
| 7 | Cost & Duration | Longer and more expensive due to de novo development and extensive trials. | Shorter and less costly because it leverages existing data from the reference biologic |
| 8 | Immunogenicity Assessment | Required but exploratory, part of overall safety evaluation. | Critical focus to ensure similarity with the reference biologic. |
| 9 | Market Approval | First-in-class approval sets the standard for future biosimilars. | Dependent on reference biologic; cannot be approved until reference is approved. |

A CRO supports sponsors across the entire clinical development lifecycle of biological and biosimilar products. This includes study design aligned with regulatory expectations, site selection, patient recruitment, bioanalytical coordination, immunogenicity assessment, data management, safety reporting, and regulatory documentation. For biosimilars, CROs also help ensure that trials demonstrate similarity to the reference product in terms of safety, efficacy, and immunogenicity.
Biosimilar clinical trials focus on proving comparability with an already approved reference biologic rather than establishing de novo efficacy. These studies emphasize analytical similarity, pharmacokinetics (PK), pharmacodynamics (PD), and immunogenicity, often with streamlined clinical efficacy requirements. In contrast, biologic trials require full clinical development to establish safety and efficacy independently.
Biological and biosimilar trials are governed by international guidelines such as ICH Q5E, ICH E6 (GCP), ICH E8, and region-specific regulations from authorities like the US FDA, EMA, and CDSCO. Biosimilars must also comply with comparability and extrapolation principles defined by these agencies, including post-marketing pharmacovigilance requirements.
Immunogenicity assessment evaluates the immune response generated by a biological or biosimilar product. Even minor manufacturing differences can impact immune reactions, potentially affecting safety and efficacy. Regulatory agencies require robust immunogenicity data to confirm that a biosimilar does not increase risk compared to its reference product.
CROs ensure compliance by aligning trial activities with current regulatory guidelines, maintaining GCP standards, managing audits and inspections, overseeing safety reporting, and ensuring data integrity. Continuous monitoring of regulatory updates helps ensure submissions meet evolving agency expectations.
Post-marketing pharmacovigilance monitors long-term safety, rare adverse events, and immunogenicity trends in real-world use. Regulatory authorities often require risk management plans and periodic safety update reports (PSURs) to ensure ongoing benefit–risk evaluation.
Global biological trials must address diverse regulatory requirements, ethnic sensitivity, and site capability variations across regions. CROs managing global trials coordinate harmonized protocols while ensuring compliance with local regulations, ethics committees, and reporting standards.
Costs are influenced by molecule complexity, trial phase, number of study sites, patient population, bioanalytical requirements, immunogenicity testing, and regulatory submission needs. Biosimilar trials may reduce costs through abbreviated clinical programs if analytical similarity is well established.
Bringing a new therapy, device, or health product to market isn’t just about research; it’s about reducing risk, accelerating timelines, and ensuring regulatory confidence. With full-spectrum clinical trial solutions, we help business leaders turn ambitious concepts into market-ready innovations while safeguarding quality and compliance.

Be the first to know the latest trends in clinical research, real-world case studies, and industry secrets.

ProRelix Research is the rapidly growing Contract/ Clinical Research Organization (CRO) with multi-country service capability supporting phase 1, 2, 3, & 4 clinical trials of Pharma, Biotech, Biopharma, Medical Device, Nutraceutical & Herbal companies to conduct in the USA, India, Europe & South East Asia.




