




Clinical trials form the decisions for regulatory approval of drugs and medical devices and as a result are the most important step of a drug development program. In addition to being lengthy and costly, clinical trials involve human subjects and therefore need to be conducted with the utmost care-keeping principles such as equity, well-being, and safety in mind. Along with the actual efficacy and safety of the new drug or device, how the clinical trial is carried out, and by whom is critical. Therefore, various factors play an important role in safeguarding the well-being of subjects in the trial: effectiveness of the intervention, risk profile, site, and personnel qualifications, the statistical design of the protocol and interpretation of results, and motivation for the trial. Adherence to Good Clinical Practice (GCP) guidelines is necessary regardless of where the trial is done and the responsible party.
What are investigator-initiated trials (IITs)?
Investigator-initiated trials (IITs) are clinical research studies that are initiated, designed and conducted by investigators from a clinical research organization (CRO). These trials are typically initiated by the investigator’s own interest in a particular topic or area of medicine. The goal of IITs is to provide evidence-based answers to important questions in healthcare.
When we think of clinical trials, we generally associate them with big pharma companies. In this case, pharma companies act as the sponsor of the trials. Sponsors initiate and take responsibility for trials that are conducted by investigators who overlook the day-to-day operations. Pharma companies or government institutions who play the role of sponsors are therefore, financially and business-purpose driven. On the contrary, investigator-initiated trials (IITs) are developed and sponsored by independent investigators or academic sponsors without participation from pharma companies. The advantages of IITs are evident and include lack of monetary purpose which reduces the chances of bias, wider therapeutic applications, and access as the trial is no longer limited by the purpose of drug companies to a few therapeutic areas that are of interest to them to increase their profits, and research at the grassroots level in laboratories or academic institutions by experts in their fields that have experience in understanding results and patient perspectives. It is expected that IITs also are faster at gathering proof-of-concept data than traditional sponsor-initiated trials which help in making decisions for the continuation or termination of clinical trials without wastage of resources. IITs can be initiated for testing new treatments as well as conducting trials to determine new therapeutic applications for already approved products expanding treatment applications to wider groups of patients. Pharma companies can provide drugs, comparators, and financial resources for the conduct of IITs or IITs may be funded by academic grants. Despite their advantages over industry-sponsored trials, IITs often suffer from financial constraints, reduced adherence to ethical considerations which is less stringent compared to sponsored trials, and
The most important individual for IITs is the sponsor-investigator. According to 21 CFR Part 312.3, a sponsor-investigator means an individual who both initiates and conducts an investigation and under whose immediate direction the investigational drug is administered or dispensed. The sponsor-investigator has both administrative responsibilities and also serves as the scientific lead for the conduct of the study. This is in contrast to most industry-sponsored trials wherein the company serves as the sponsor to initiate the trial and investigators from academic or medical institutions carry out the trial operations.
Regulatory considerations for IITs
Regulatory approval and steps for IITs are the same as they are for sponsored trials and involve the following aspects as shown in Figure 1. The overarching objective is to ensure that trials are conducted ethically, responsibly, and with a strong scientific background keeping subject safety at the apex.
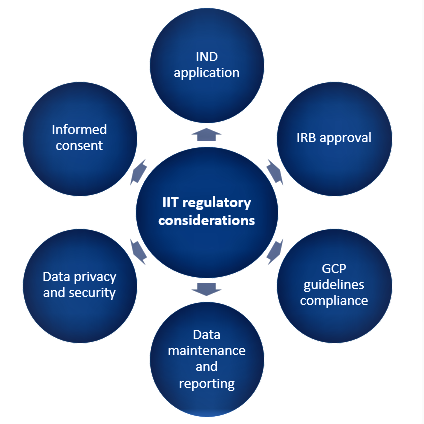
Figure 1. Aspects for regulatory considerations for IITs
Investigational New Drug (IND) submission:
This includes sponsor-investigator information, investigator’s brochure (not mandatory but recommended), clinical trial protocol, chemistry, manufacturing, and control (CMC) information, pharmacology, and toxicology information, and summary of previous human experience (if any). In certain cases, a sponsor-investigator may be exempt from IND submission, the IND may be abbreviated, and in some cases cross-referenced to a commercial IND if the drug is already authorized. The forms required for an IND submission include FDA Form 1571 Investigational New Drug Application, FDA Form 1572 Statement of Investigator, and Form FDA 3674 Certification of Compliance. The IND review process by the FDA involves a 30-day period following receipt of the IND application wherein either the clinical trial can proceed, or a clinical hold is placed which requires a response by the sponsor-investigator followed by re-review by the FDA.
The Investigational New Drug (IND) submission is an essential step in the clinical trial process. IND clinical trial involves submitting a detailed application to the FDA that outlines the proposed clinical trial design, patient population, and safety assessment. This application must be completed before any clinical trials can begin. The IND submission is a critical part of ensuring that all trials are conducted safely and ethically. By providing the FDA with detailed information about the proposed trial, they can ensure that all risks are minimized and that patients receive quality care throughout the trial process.
Institutional Review Board (IRB) approval:
IRB approval of the protocol is prior to the commencement of the trial. This is vital to ensure that the rights and safety of the subjects are protected and that they are not exposed to any untoward foreseeable risks. Ongoing monitoring of trial activities by the IRB is also necessary which evaluates and collects data on any adverse events that can jeopardize the subject health.
Informed consent:
Obtaining informed consent is necessary for IITs where the subjects are informed and educated about the trial drug/medical device, dosing schedules, any risks, and monitoring visits. Subjects are also given the right to withdraw from the trial at any time.
Informed consent is a critical process in healthcare industry and other research-related fields. It ensures that the patient or research participant is aware of all the risks, benefits, and alternatives associated with a particular medical procedure or study. To ensure that informed consent is obtained, Electronic Informed Consent Form(e-ICF) has been developed. This form helps to streamline the process of obtaining and documenting informed consent from patients or research participants. It also helps to ensure accuracy and consistency in the information that is provided to them. With e-ICF, healthcare providers can easily obtain informed consent from their patients in a secure manner.
Good Clinical Practice (GCP) compliance:
ITs must adhere to ICH-GCP guidelines as well as any local clinical practice guidelines to ensure subject safety and trial integrity. All personnel involved in trial activities must be trained on GCP guidelines including good documentation practices.
Good Clinical Practice (GCP) compliance is an essential requirement for any medical research or clinical trials. It is a set of principles and guidelines that ensure the safety, quality and efficacy of the research and trials conducted by healthcare professionals. A professional certificate in ICH GCP can help healthcare professionals understand the importance of GCP compliance and how to implement it in their work. It provides them with a comprehensive understanding of the principles, processes and procedures that must be followed in order to ensure compliance with GCP regulations. With this certification, they can develop an effective system for monitoring, reporting and documentation that meets regulatory requirements.
Data maintenance and reporting:
Data from the trial must be reported on time keeping in mind the requirements of good documentation practices. Adverse event reporting is essential to prevent risks to participants and must be done promptly. Important information from trials must be comprehensively recorded at the time of occurrence to ensure adherence to protocols and any changes must also be included in trial files.
Clinical data management is a critical element of the regulatory considerations for IITs. It involves the collection, storage, and analysis of clinical data to ensure that it is accurate and up-to-date. This helps to ensure that all regulatory requirements are met, as well as providing insights into the performance of the IITs. clinical data management services play an important role in ensuring that clinical data is managed effectively and efficiently.
Data privacy and security:
Sponsor-investigators in IITs are responsible for ensuring that trial data is collected, stored, and shared in compliance with regulatory requirements such as the Health Insurance Portability and Accountability Act (HIPAA) in the United States.
Study designs for IITs
IITs can be observational or interventional. The various study designs that are commonly used in IITs include:
Randomized controlled trials:
these provide the highest level of evidence and serve as the gold standard for treatment comparisons. In these types of trials, two groups are randomly assigned the intervention and the comparator (active drug or placebo) and monitored over time by physicians and study staff who are usually blinded to the treatment assignment. These trials provide the lowest level of bias.
Single-arm studies:
only one group is assigned treatment and is usually done in case of rare diseases.
Non-interventional studies:
these include primary data collection such as prospective observational studies and registries where data is collected from routine clinical care or secondary use of data which involves retrospective studies.
Adaptive trial designs:
these are becoming increasingly popular and involve change in protocol such as sample size, treatment assignment, etc. based on interim results and therefore can save money and time.
Publication strategy for IITs
In order to maximize the benefits with conducting IITs, it is important that the information that is obtained is disseminated via publications to inform healthcare decision makers and physicians on the benefits or risks associated with the experimental intervention. It is important to include data, whether positive or negative from studies in systematic reviews and meta-analysis to ensure that the decisions made are correct as leaving out studies can lead to erroneous conclusions. Study registries and prospective study registrations can help make data available publicly and help in supporting healthcare decisions as well as further research. Any publication strategy for an IIT should include the following:
- Identification of an appropriate target audience such as healthcare decision makers, physicians, patients, caregivers, etc.
- Selection of a suitable journal for publication based on publication type (peer-reviewed manuscript, editorial, poster) and therapeutic area.
- Development and submission of the manuscript as per the journal requirements for the author.
Thus, IITs form an important part of healthcare decision making as they are purported to present an unbiased view of treatments and provide access to a wider range of therapeutic options. Their widespread application is limited by funding restrictions as compared to industry-sponsored trials and as a result are currently used to provide preliminary research results to support larger trials.
References
- Investigator initiated trials versus industry sponsored trials – translation of randomized controlled trials into clinical practice (IMPACT) | BMC Medical Research Methodology | Full Text (biomedcentral.com)
- Investigational New Drug Applications Prepared and Submitted by Sponsor-Investigators | FDA
- Investigator Initiated Trials: a guide for prospective Investigators (novartis.com)
- Investigator Initiated Studies | MyAlcon.com

