




Explore 2025 global clinical trial compliances helping CROs achieve operational excellence with quality, safety, and regulatory efficiency.
Table of Contents
Abstract
The global clinical research landscape in 2025 is undergoing a transformative shift. New regulatory frameworks, such as ICH E6(R3), updated FDA guidance on decentralized trials, and the full operationalization of the EU Clinical Trials Regulation (CTR), are redefining what operational excellence means for Contract Research Organizations (CROs). This review explores the latest global trial guidelines that influence CRO operations, highlights the implications of these evolving standards, and outlines practical strategies that enable CROs to achieve quality, efficiency, and compliance in an increasingly complex environment.
Introduction
Clinical research organizations have long been central to the successful execution of global clinical trials. However, the operational frameworks that guide these organizations are evolving rapidly. The year 2025 represents a pivotal moment, as regulatory bodies across regions are aligning around shared principles of quality by design, risk-based management, transparency, and patient-centricity.
The newly finalized ICH E6(R3) guideline is a landmark update that redefines Good Clinical Practice (GCP) in a way that accommodates modern study designs, real-world data, and digital technologies. Simultaneously, the U.S. Food and Drug Administration (FDA) has expanded its guidance for decentralized clinical trials (DCTs), emphasizing oversight, data integrity, and participant safety. In the European Union, the Clinical Trials Information System (CTIS) under the CTR has become fully operational, driving harmonization and transparency.
For CROs, these changes represent both a challenge and an opportunity. The challenge lies in adapting operations, systems, and workforce skills to meet these new expectations. The opportunity lies in leveraging these guidelines to enhance operational efficiency, strengthen sponsor trust, and drive global competitiveness.
Key Global Regulatory Developments
ICH E6(R3): The Reimagined Good Clinical Practice
The International Council for Harmonisation (ICH) officially adopted E6(R3) in early 2025. This updated guideline replaces the older prescriptive approach with a more principle-based framework. Its goal is to ensure that GCP remains relevant in the context of diverse trial designs, including adaptive, hybrid, and decentralized studies.
The key themes of E6(R3) include quality by design (QbD), risk-proportionate management, and evidence-based decision-making throughout the clinical trial lifecycle. Instead of treating GCP as a rigid checklist, E6(R3) encourages sponsors and CROs to apply critical thinking to determine which processes are essential to data integrity and participant protection.
CROs must now ensure that every trial design incorporates quality at the earliest planning stages, that risk assessment is continuous, and that critical-to-quality (CtQ) factors are defined and monitored through fit-for-purpose controls.

FDA Guidance on Decentralized and Hybrid Clinical Trials
The FDA has been proactive in aligning with global modernization efforts. The latest guidance on decentralized clinical trials emphasizes maintaining patient safety and data integrity when trial activities occur outside traditional research sites. Remote assessments, telemedicine, home-based sample collection, and the use of digital health devices are now recognized as valid components of trial design when supported by clear documentation and oversight. CROs are expected to establish strong data provenance systems, ensure proper vendor qualification, and document delegation of trial responsibilities. Importantly, E6(R3) principles are reflected in the FDA’s stance that sponsor oversight cannot be outsourced, even when CROs and third-party providers execute decentralized elements.
EU Clinical Trials Regulation (CTR) and CTIS
The European Medicines Agency (EMA) achieved full implementation of the Clinical Trials Regulation (EU No 536/2014) and its supporting Clinical Trials Information System (CTIS) in 2025. All new clinical trials conducted in the EU must now be submitted, managed, and reported through CTIS.
For CROs, this means adapting to a single-entry system for submissions and transparency, managing country-specific timelines, and ensuring data consistency across public disclosures. CTIS requires CROs to maintain comprehensive documentation and communication systems that integrate regulatory, operational, and data management workflows.
Table 1: Summary of Key CTIS Compliance Milestones for CROs
| Sr.No | Milestone | Implementation Timeline | Key Requirements for CROs | Operational Impact |
| 1 | Full Transition to CTIS | January 31, 2025 | All new and ongoing clinical trials in the EU must be managed exclusively via the Clinical Trials Information System (CTIS). | CROs must ensure all submissions, communications, and trial updates occur through CTIS. Legacy EudraCT processes are no longer accepted. |
| 2 | Unified Application Submission | Effective from 2025 | Single application dossier submission for all EU Member States concerned. | Simplifies multi-country submissions but requires harmonized documentation and real-time collaboration across regions. |
| 3 | Transparency and Public Disclosure | Continuous | Obligation to redact commercially confidential information (CCI) and personal data before publication on CTIS public portal. | CROs must implement document redaction processes and designate responsible compliance officers. |
| 4 | Regulatory Timelines Enforcement | From 2025 onward | Strict adherence to statutory timelines for review, response, and approval under CTR framework. | CROs must update project management systems to track and meet regulatory deadlines accurately. |
| 5 | Safety Reporting Integration | Phased integration through 2025 | Integration of serious adverse event (SAE) and SUSAR reporting workflows with CTIS and EudraVigilance. | Ensures synchronized safety reporting and real-time regulatory visibility into safety data. |
| 6 | User Role and Access Management | Ongoing (2025–2026) | CROs must define clear roles for sponsor, clinical project managers, and regulatory users within CTIS. | Establishes accountability, reduces access-related compliance risks, and improves audit traceability. |
| 7 | Training and System Familiarization | Continuous (2024–2025) | Mandatory training on CTIS modules, submission workflows, and change control for CRO staff. | Builds internal expertise, minimizes submission errors, and improves regulatory communication. |
WHO and Global Ethics Harmonization
The World Health Organization (WHO) released updated guidance in 2024 and 2025 promoting best practices for ethical trial conduct, particularly in low- and middle-income countries. These guidelines emphasize research preparedness, ethical oversight, and capacity-building to strengthen global trial ecosystems.
CROs conducting multinational studies must align their practices with WHO-recommended standards for informed consent, data sharing, and post-trial access to treatments. This ensures ethical consistency across global study networks.
Diversity, Equity, and Inclusion (DEI) Requirements
Regulatory agencies, particularly in the United States, are increasingly focused on ensuring diversity in clinical trial participation. The FDA’s recent policy initiatives encourage sponsors and CROs to develop diversity action plans that outline recruitment strategies, community engagement, and data transparency for underrepresented populations.
CROs that can demonstrate meaningful inclusion efforts and community partnerships will not only comply with regulatory expectations but also build stronger trust with sponsors and patient groups.
Operational Implications for CROs
The new regulatory environment directly affects how CROs must plan, execute, and monitor clinical trials. Operational excellence in 2025 is no longer defined by speed and cost alone, but by the ability to deliver high-quality, compliant, and patient-centered trials with measurable efficiency.
Quality by Design and Risk-Based Oversight
Every trial must now be built on a foundation of quality by design. CROs should conduct cross-functional QbD workshops to identify critical-to-quality factors and define proportionate controls. This approach ensures that resources are focused on processes and data that matter most to participant safety and scientific validity.
Risk-based monitoring (RBM) models have matured into hybrid systems combining centralized analytics and on-site verification. CROs must integrate statistical data monitoring tools capable of detecting outliers and trends in real time.
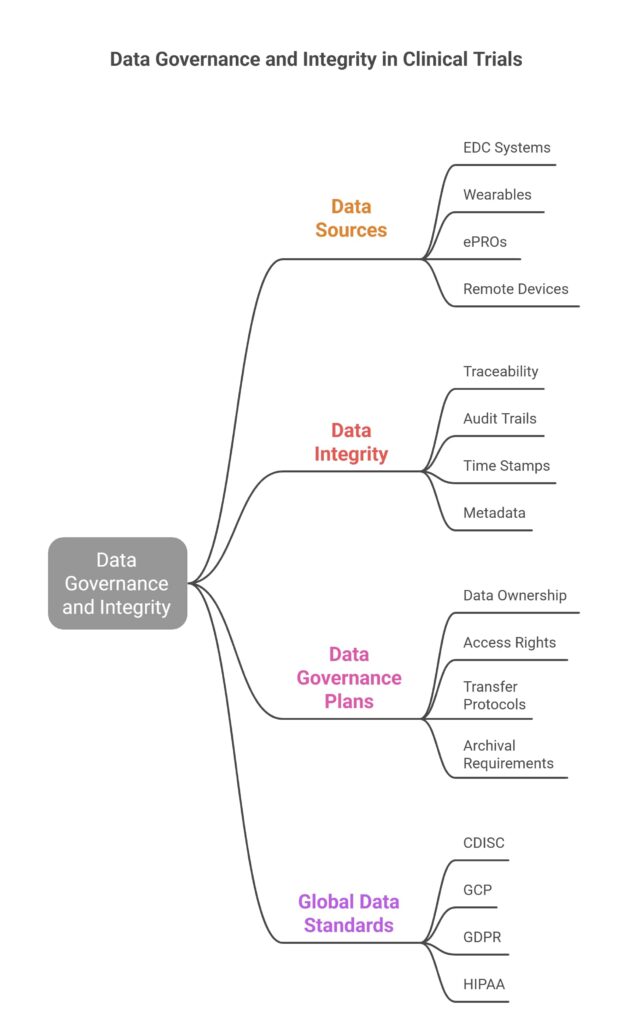
Data Governance and Integrity
With digital data streams coming from EDC systems, wearables, ePROs, and remote devices, CROs must ensure end-to-end data integrity. Each data point must be traceable to its source with verifiable audit trails, time stamps, and metadata.
Data governance plans must specify data ownership, access rights, transfer protocols, and archival requirements in compliance with GCP, GDPR, and HIPAA standards. Adopting global data standards such as CDISC can streamline submissions and facilitate regulatory review.
Data Flow and Provenance Map for Multisource Clinical Trial Data:
Vendor Oversight and Accountability
Decentralized models often involve a wider network of vendors, including home healthcare providers, laboratory logistics companies, and telemedicine partners. CROs must maintain structured vendor qualification processes, periodic audits, and clearly defined service-level agreements.
Vendor oversight dashboards that monitor performance indicators, audit findings, and data transfer reliability are becoming an essential tool for CRO quality management systems.
Patient-Centric Trial Design
A patient-centric approach is at the heart of modern guidelines. CROs should promote the use of simplified consent forms, flexible visit schedules, and remote engagement strategies that reduce participant burden.
CROs are also expected to evaluate and document barriers to participation for specific populations, ensuring equitable access. Training site staff in cultural sensitivity and using multilingual study materials can further enhance inclusivity.
CTIS and Regulatory Submission Readiness
Operational readiness for CTIS submissions requires updated SOPs and digital infrastructure. CROs must develop a structured document management system that supports electronic submission, regulatory correspondence, and version control.
Timely compliance with the CTR’s transparency requirements also means planning for redaction of commercially confidential information before public disclosure.
Workforce Training and Competency Development
The success of these new frameworks depends heavily on human capability. CROs should implement role-based training programs to build competencies in quality by design, decentralized trial management, risk-based monitoring, and data governance.
Cross-functional collaboration among clinical operations, data management, biostatistics, and quality assurance teams ensures consistent interpretation of regulatory expectations. Many CROs are now developing “Regulatory Readiness Teams” responsible for continuous monitoring of global guideline updates and translating them into actionable operational SOPs.
Table 2: Recommended Role-Based Competency Areas for CRO Staff under ICH E6(R3)
| Sr.No | Role | Core Competency Areas | Description / Key Skills | Training Frequency |
| 1 | Clinical Research Associate (CRA) | GCP and ICH E6(R3) Compliance, Monitoring Practices, Risk-Based Monitoring (RBM), Data Integrity, and Informed Consent Process | CRAs should ensure that trial sites comply with protocol and regulations, use remote and on-site monitoring effectively, and document findings accurately. | Annual or as per regulatory update |
| 2 | Clinical Trial Manager (CTM) | Project Management, Site Oversight, Risk Assessment, Vendor Coordination, Quality Assurance | CTMs oversee overall project timelines, ensure compliance, and mitigate risks based on early signal detection. | Annual plus role transition training |
| 3 | Regulatory Affairs Specialist | Global Regulatory Guidelines (FDA, EMA, MHRA, DCGI), CTIS Portal Operations, Submission Readiness, Ethical Compliance | Ensures submission accuracy, maintains CTIS timelines, and manages interactions with competent authorities. | Biennial or upon regulatory updates |
| 4 | Data Manager | Data Integrity, eCRF Design, Source Data Verification, Audit Trail Management, GDPR and HIPAA Compliance | Responsible for clean, accurate, and validated trial data with compliance to data protection laws. | Annual |
| 5 | Quality Assurance (QA) Auditor | Internal Audit Skills, CAPA Implementation, Inspection Readiness, GCP Auditing | Conducts periodic quality audits and ensures continuous improvement in operational processes. | Annual |
| 6 | Medical Monitor / Safety Officer | Pharmacovigilance, SAE/AE Reporting, Signal Detection, Safety Data Review | Oversees safety data, performs causality assessments, and ensures compliance with ICH E2A and E6(R3). | Annual or per safety system update |
| 7 | Biostatistician | Statistical Analysis Plan (SAP), Risk-Based Design, Data Visualization, Trial Endpoints Interpretation | Ensures that trial design and data analysis comply with regulatory and scientific standards. | Every two years |
| 8 | Clinical Trial Assistant (CTA) | Documentation Management, TMF Maintenance, Communication Coordination, Administrative Support | Assists operational teams in maintaining documentation integrity and communication flow. | Annual |
| 9 | Investigator / Site Staff | Protocol Adherence, Patient Safety, Informed Consent, EDC Data Entry Accuracy | Ensures patient rights, safety, and well-being while maintaining accurate site data. | Initial GCP training + refreshers every 2 years |
Technology and Infrastructure Investments
Modern CROs must evolve technologically to remain compliant and competitive. Key investments include:
- Integrated Data Platforms: Systems that can aggregate and normalize data from multiple sources, providing real-time insights into study performance.
- Centralized Monitoring Analytics: Machine learning tools for detecting protocol deviations, data anomalies, and site performance trends.
- Secure Remote Access Systems: Compliant identity management and encryption solutions for remote monitoring and telehealth visits.
- Regulatory Submission Tools: Automated CTIS dossier builders and redaction software to streamline submission workflows.
These tools not only improve regulatory compliance but also enhance efficiency and data-driven decision-making.
Implementation Challenges and Mitigation Strategies
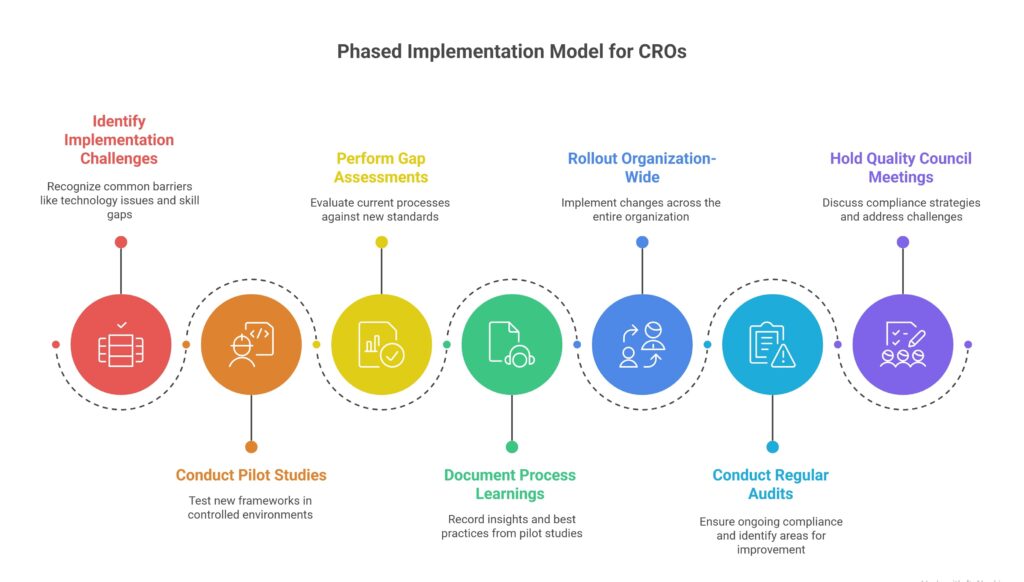
Despite clear regulatory guidance, CROs often face practical challenges in execution. Common barriers include technology integration issues, variable regional interpretations of guidelines, and skill gaps within operational teams.
To overcome these challenges, CROs can adopt a phased implementation model. This includes conducting pilot studies under the new frameworks, performing E6(R3) gap assessments, and documenting process learnings before organization-wide rollout. Regular internal audits and quality council meetings help maintain compliance momentum.
The Road Ahead: Building Sustainable Operational Excellence
Looking forward, operational excellence will depend on how effectively CROs embed regulatory compliance within their culture and systems. The future CRO model will likely combine automation, data transparency, and adaptive quality management to handle increasingly complex global trials.
The key is not just compliance, but demonstrable assurance—the ability to show regulators and sponsors that decisions are evidence-based, risks are proactively managed, and data integrity is uncompromised.
CROs that invest early in digital transformation, integrated oversight, and workforce development will be better positioned to thrive under the evolving 2025–2030 regulatory landscape.
8. Conclusion
The 2025 global trial guidelines collectively represent a paradigm shift in how clinical research is conducted and managed. The transition from prescriptive checklists to principle-driven frameworks demands that CROs evolve from task executors to strategic partners in trial design and oversight.
ICH E6(R3) serves as the foundation for this transformation, complemented by FDA’s decentralized trial guidance, the EU CTR/CTIS framework, and global calls for ethical inclusion and data transparency. Achieving operational excellence now means embedding quality, adaptability, and patient-centeredness into every layer of the CRO organization.
CROs that embrace this transformation not only ensure compliance but also build the trust, resilience, and innovation capacity required for the next era of clinical research.
Read More: Faster Drug Development with Basket, Umbrella, and Platform Trial Strategies
Frequently Asked Questions (FAQs)
How does ICH E6(R3) differ from ICH E6(R2)?
ICH E6(R3) focuses on flexibility, risk-based oversight, and digital data management, moving beyond the rigid compliance model of E6(R2).
What is the importance of CTIS for CROs?
CTIS ensures transparency, centralized submissions, and compliance across EU member states, reducing delays in trial approvals.
How can CROs achieve operational excellence in 2025?
By adopting advanced data systems, continuous staff training, and robust quality management aligned with ICH E6(R3).
What benefits do the new guidelines bring to global collaboration?
They promote unified documentation, digital integration, and regulatory consistency for smoother multinational trial execution.
References
- International Council for Harmonisation. ICH E6(R3): Guideline for Good Clinical Practice. Final version adopted 06 Jan 2025. Available at: https://database.ich.org/sites/default/files/ICH_E6%28R3%29_Step4_FinalGuideline_2025_0106.pdf ICH Database+1
- U.S. Food and Drug Administration. Guidance for Industry: Conducting Clinical Trials With Decentralized Elements. October 16 2025. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/conducting-clinical-trials-decentralized-elements U.S. Food and Drug Administration+1
- U.S. Food and Drug Administration. ICH E6(R3) Good Clinical Practice (GCP): Guidance for Industry. September 8 2025. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e6r3-good-clinical-practice-gcp U.S. Food and Drug Administration
- European Medicines Agency. ICH E6(R3) Guideline for Good Clinical Practice (GCP). Step 5 document. January 23 2025. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e6-r3-guideline-good-clinical-practice-gcp-step-5_en.pdf European Medicines Agency (EMA)
- Summary of key changes in ICH E6(R3). Available at: https://www.ct-toolkit.ac.uk/news/summary-key-changes-ich-e6-r3-guidelines ct-toolkit.ac.uk
