The COVID-19 pandemic has transformed the worldwide regulator performing on approval of medicine as a fast-track process and involved in mutual accord in bringing a drug of importance for the civic. This is a major challenge as the safety and effectiveness of novel therapies are not fully established but ways to bring about a difference to the COVID-19 pandemic response is the need of the hour. Regulators have been go-getting hard in order to bring flexibility in the regulations so as to quicken various approval and adaptive licensing mechanisms. Various institutions have been working together in an alliance to fight this pandemic. The WHO has always promoted such cooperative approaches as these enhance the efficiency of regulatory authorities and also avoids copying. We have focused on details of clinical trials and diverse classes of drugs that are suitable to treat COVID 19 infection.
The nodal agency for the evaluation of safety and efficacy of marketed medicines in the USA is FDA. The Center for Drug Evaluation and Research under FDA reviews drugs, post-marketing surveillance, etc. Pharmaceutical companies will apply for Investigational New Drug (IND) to FDA before commercial marketing for its approval. The application of IND can be investigator IND, treatment IND, or emergency use IND. Investigator IND proposes approved or unapproved drugs for new patient populations or new indications. Emergency use IND permits for emergency use of the experimental pharmaceutical product in an emergency like the COVID-19 pandemic. Treatment IND permits in case of serious or immediately life-threatening conditions.
In New Drug Application (NDA), IND data from animal and human studies will be submitted for approval. NDA application is critically reviewed by FDA reviewers for safety and efficacy in a proposed use, risk-benefit analysis, appropriateness of package inserts, and quality and purity of drug during manufacturing, maintenance, and preservation. The generic drug product receives approval under Abbreviated ANDA. The generic drug product can be safe and effective or a low-cost alternative for the intended disease and marketed with an alternative brand name. Therapeutic biologics application deals with cytokines, monoclonal antibodies, enzymes, thrombolytics, growth factors, immunomodulators, animal-derived proteins, or their recombinant version and other nonvaccine therapeutic immunotherapies. Expedited approval mechanisms for new drug products: in this category, pharmaceutical companies can propose their product under various categories for faster approval for drugs. Different mechanisms for faster approval of drugs are discussed below.
Fast track procedure
Under this, the development and review of drugs is expedited for the treatment of serious or unmet medical conditions. This process enables access to new drugs to the patient. An unmet medical need is defined as “providing a therapy where none exists or providing a therapy which may be potentially better than available therapy,” Drugs under this category will conduct clinical trials using specific biomarkers to prove their efficacy.
Breakthrough therapy
Under this category, drug products fill the gap in the treatment of a serious condition designed for development. The efficacy of these products is derived from the early clinical trial outcomes related to clinically significant endpoints. The drug products under this category receive benefits of fast-track approval along with senior mentors of the FDA from the Phase 1 trial.
Accelerated approval
Under this pathway, a surrogate endpoint is used instead of clinical endpoints to fill an unmet medical need for serious conditions. The pharmaceutical agents who received approval under this category need to complete phase 4 confirmatory trials to the actual benefit for which the drug was approved. In 2020, FDA proposed the withdrawal of hydroxyprogesterone caproate injection from the market for the prevention of recurrent preterm birth. The neonatal outcomes in the treatment group showed that there was no significant improvement.
Priority review
This shortens the duration of the FDA’s review. Priority review is given for serious conditions for which the pharmaceutical product is shown to be safe and effective from the available treatment.
The USFDA along with NIH is involved in Accelerating COVID-19 Therapeutic Interventions and Vaccines for the development of therapeutic candidates and vaccines that can help end the COVID-19 global pandemic.
Table 1– Novel drug approvals for 2020

A statutory requirement for emergency use authorizations
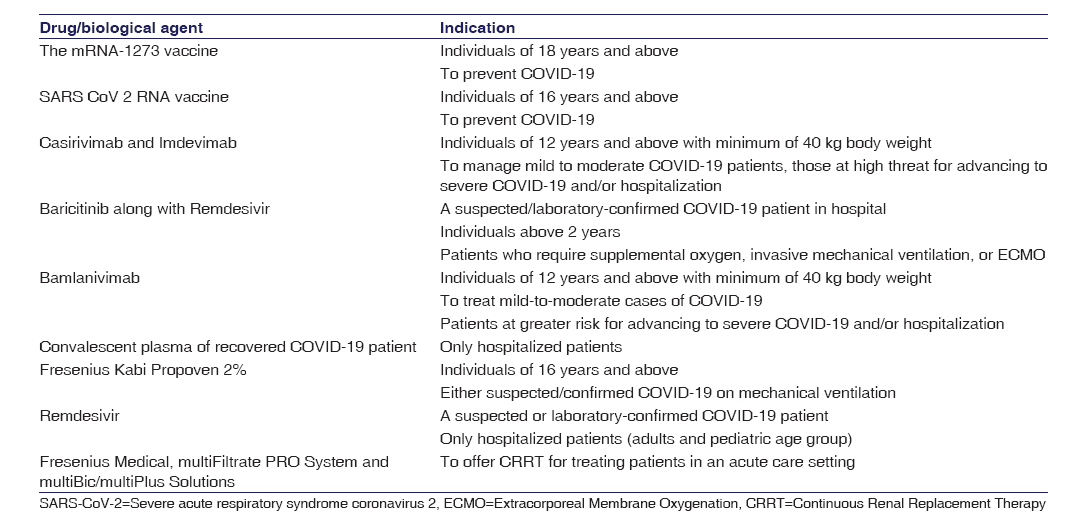
This is granted when a medical product shows effectiveness in serious or life-threatening diseases. While authorizing emergency use for any product, the probable benefit for the proposed use should outweigh its recognized and potential hazards. As of December 18, 2020, total in vitro diagnostic products was 305 (molecular tests and sample collection devices-233, antibody tests-62, and antigen tests-10), Personal Protective Equipment and Related Devices were 24, and >100 Ventilators and accessories, and Drug and Biological Products were 10. In [Table 1], details of drugs and biologics approved by the USFDA under Emergency Use Authorizations (EUA) are mentioned and in [Table 2] all the COVID – 19 Emergency Use Authorization treatments are described. Shuren and Stenzel observed that molecular diagnostic tests for COVID-19 that received approval under EUA was a reasonable step in accessing diagnostic steps during emergency situation-like the COVID-19 pandemic. Initiatives such as international collaboration, large scale production of small number of well designed, well-developed, and validated tests along with the reliable methods of validation and its test performance are essential for molecular diagnostic test to beat a pandemic like COVID-19.
Hydroxychloroquine sulfate (HCQ) and Chloroquine phosphate (CQ) received EUA for the treatment of COVID-19. However, a scientific review of CQ and HCQ observed that potential benefits outweigh the known (serious cardiac adverse events) and other potential adverse effects.
Table 2– Drug and biological products approved by US Food and Drugs Act under emergency use authorization