




Foreign Clinical Trials (FCTs)
Prior to the 1962 Kefauver-Harrison Amendments, it was uncommon for a Sponsor to submit FCTs data. In 1975 provisions that permit the submission of FCTs data not conducted under an IND were codified in 21CFR312.120. Since then, medical product discovery & development have become increasingly globalized. In the past three decades, drug development, production & distribution moved from a national and regionally‐ centered activity to a global one. This increase in shift has significant implications on FDA’s perspective on international clinical trials.
International Clinical Investigators & Clinical Trials
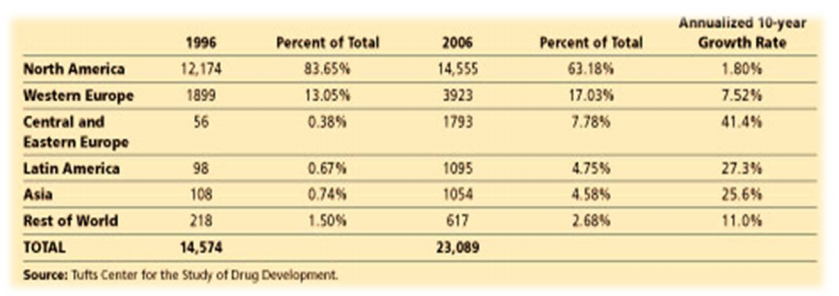
Figure 1 – Rate of Increase for FDA Regulated Investigators: 1996-2006
During the 10-year period(1996 to 2006), the % of the total for U.S. based investigators declined by 20% (annual growth rate 1.8%). The number of active FDA–regulated investigators who are based outside the United States has grown.
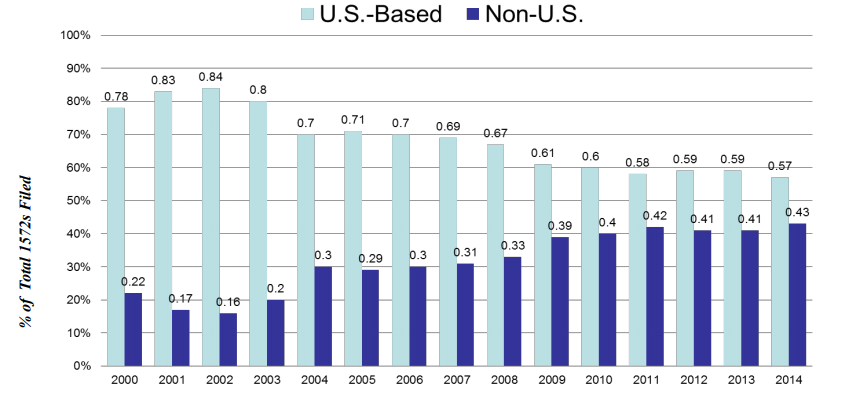
Figure 2 – Rate of Increase for FDA Regulated Investigators: 2000-2014
OIG Report:

Over half of all clinical trial sites are outside the U.S. % of non -U.S. clinical investigators conducting trials under INDs has doubled over the last decade. 80 % of applications for drugs & biologics contain data from ex -U.S. studies. 78 % of all subjects were enrolled outside the U.S. 87 % of all subjects in recent biologics trials were enrolled outside the U.S.
Distribution of Studies
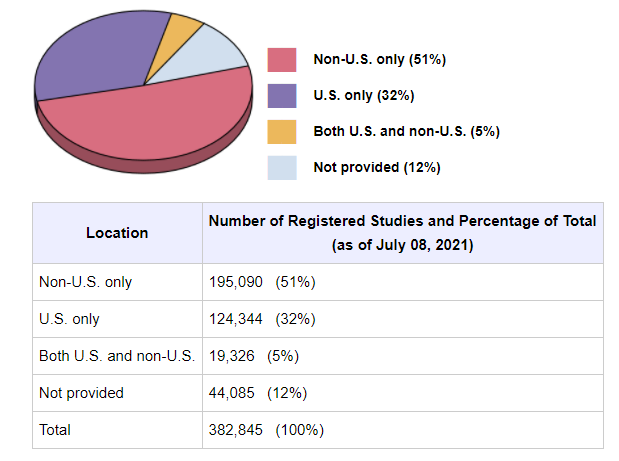
Figure 3 – All studies registered on ClinicalTrials.gov: Data as of July 08, 2021
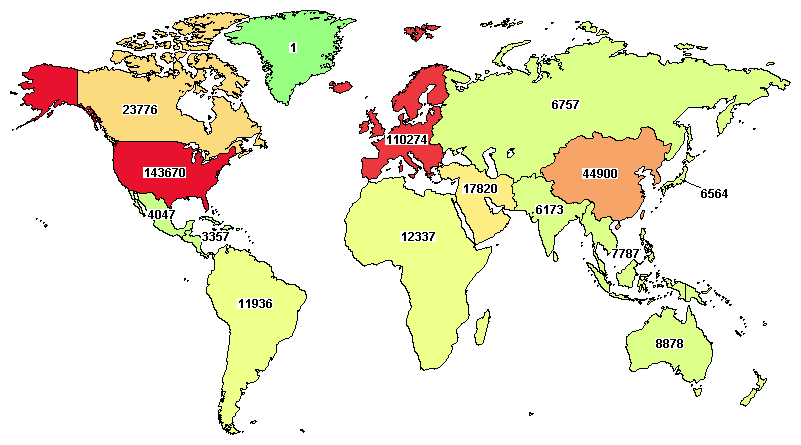
Figure 4 – ClinicalTrials.gov currently lists 174,156 studies with locations in 187 countries
What Drives Companies to Conduct Clinical Trials Outside the USA?
Pushing & Pulling Forces
US | NON – US |
| · Difficulty recruiting patients – Fewer feel need to participate · Higher standard of care · Mistrust (historical infamy Tuskegee trial) · Requirement for larger # of Pts · Insurance precluding participation/HIPAA · Higher costs: clinical care/Operational/ documentation & training) | · Reduced cost or economic drivers (clinical care/Operational/training) · Motivated subjects & investigators · Rx naïve subjects willing & eager to participate · Faster recruitment rates/ short timeline · Availability of large # of patients · Countries and institutions trying to attract research and clinical trials · Availability of CROs focused on global trials · Less regulatory red tape |
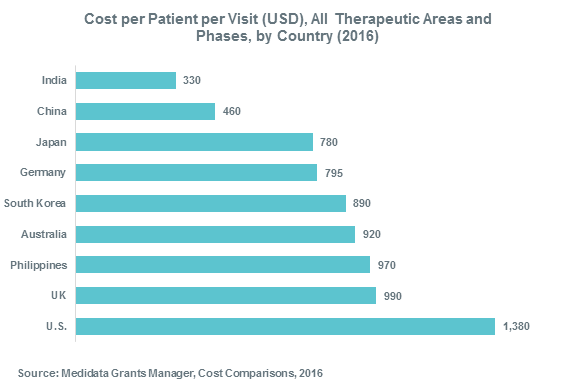
Figure 5
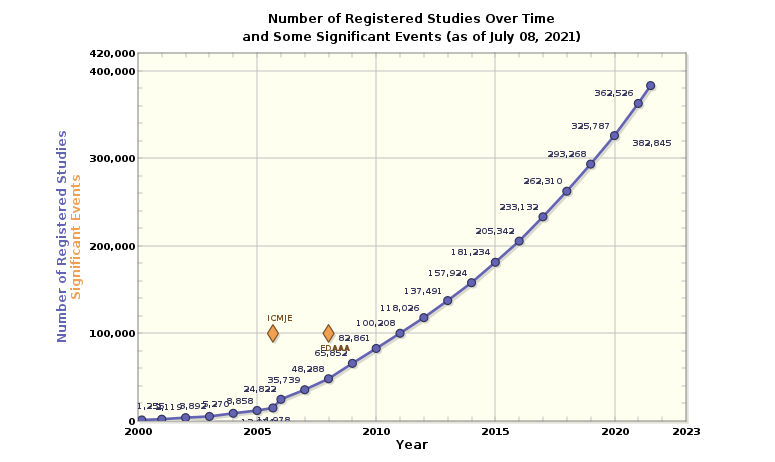
Figure 6 – Number of Registered Studies Over Time
Current Law Governing Foreign Clinical Studies:
- Foreign Study Conducted Under IND Application.
When a study is conducted under an IND but is located outside of the United States, the study still must comport with all relevant FDA regulations as if it were being conducted within the United States. A Sponsor is not required to conduct an FCT under an IND in order to use it as support for an NDA or IND.
- Foreign Clinical Trials Not Conducted Under an IND.
Acceptance of Foreign Clinical Data: FDA Accepts Foreign Clinical data from studies not conducted under an IND if the following conditions are met:
- Study was conducted in accordance with Good Clinical Practice (GCP).
- FDA is able to validate the data from the study through an onsite inspection.
GCP:
Sponsor shall submit to FDA a description of actions taken to ensure research conformed to GCP: – Defined as a standard for the design, conduct, performance, monitoring, auditing, recording, analysis, and reporting of clinical trials in a way that provides assurance that the data &reported results are credible and accurate and that the rights, safety, and well-being of trial subjects are protected.
Supporting Information:
- CI’s qualifications.
- Description of the research facilities.
- Summary of the protocol & results of the study &, should FDA request, case records
- Description & details of the product/drug used.
- Information showing that the study is adequate & well controlled.
- Name & address of the IEC, a summary of the IEC’s decision.
- A description: – how IC was obtained – what incentives, if any, were provided to subjects in the study – how the sponsor(s) monitored the study – how CIs were trained to comply with GCP.
Validation of Data: Onsite Inspections
- Help to ensure the protection of rights, safety and welfare of research participants.
- Help to ensure data that are submitted in marketing applications are fit for the purpose
For regulatory decision making (Approval)
As the evidence base for clinical use of the drug (Label)
- Allow evaluation of compliance with FDA regulations.
Assigning International Inspections
Clinical sites likely to be audited when: – only foreign data are submitted to support an application – insufficient domestic data – domestic and foreign data show conflicting results pertinent to decision-making – there is a serious issue to resolve (e.g., suspicion of fraud, scientific misconduct, significant human subject protection violations).
Useful Guidance
FDA Acceptance of Foreign Clinical Studies Not Conducted Under an IND Frequently Asked Questions – Issued March 2012 – Provides clarifications for sponsors and applicants on how to demonstrate compliance with the requirements of 21 CFR 312.120.
The challenges
- Increase in Breadth of Inspections.
- Increasing volume to data from other regions submitted in support of marketing submissions submitted to FDA.
- Increase in the breadth of international inspections.
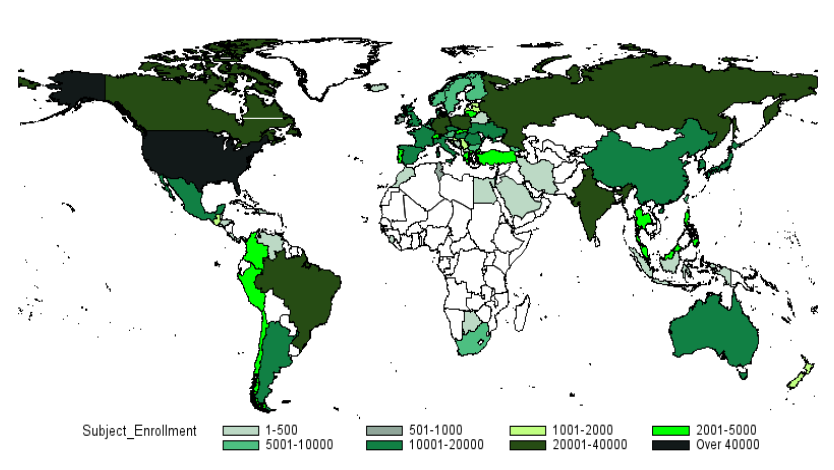
Figure 7 – Distribution of Subject Enrollment
Finite inspection resources:
The breadth of international inspections coupled with finite inspection resources results in the inspection of a limited number of sites. Despite the large portion of clinical studies being conducted abroad, the OIG 2010 Report found that the FDA only inspected 0.7% of all foreign clinical sites in 2008, compared to 1.9% of all domestic clinical sites.
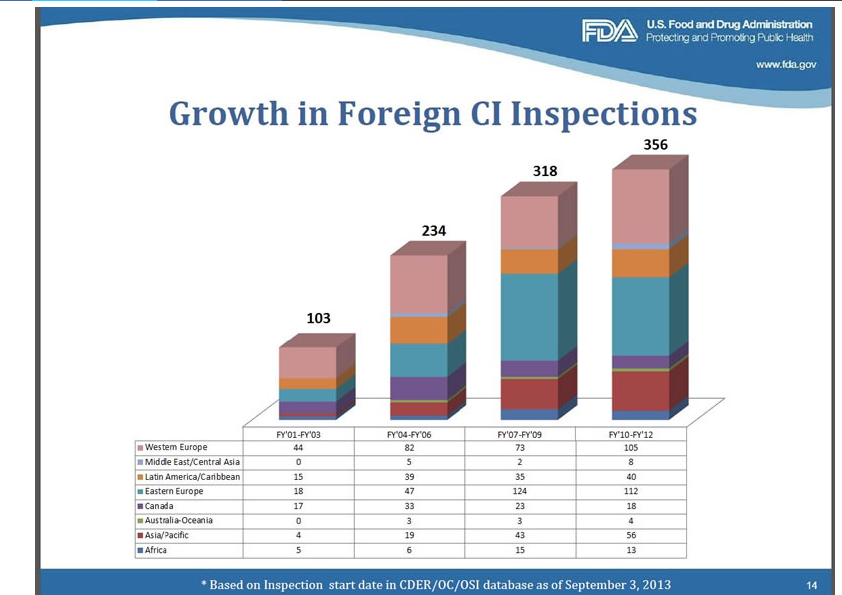
Figure 8
Strategies
Leveraging Knowledge/ Resources:
- Utilize finite resources strategically and efficiently and leverage inspection – EMA-FDA Collaboration GCP Inspection Initiative
- Initiated in Sept. 2009 as a pilot to
- Develop a joint GCP program – Plan, coordinate, schedule, and conduct collaborative GCP inspections
- Establish joint procedures – Share information related to drug applications – FDA International Offices:
- Provide a platform for inspection of foreign facilities.
- Offices collect & leverage local & regional knowledge.
Building Quality:
Build quality during the planning stages: – incorporate quality management approach into clinical studies & their oversight to prevent the critical/errors, and monitor for those errors (perhaps centrally (remotely/real-time)) and focus less on the less important matters.
Utilize Data Standards:
- Encouraging sponsors to utilize data standards to improve analysis and site selection for inspection.
- Risk-Based Monitoring and Inspection – Using innovative strategies & tools to identify sites for clinical inspection(ex. GCP risk-based site selection tool for clinical inspection).
Science-Based Standards:
- Working to converge regulatory standards (to share a common foundation of science-based goals for product safety, quality, and efficacy). – Continue to work to converge regulatory standards, processes, and procedures for the pharmaceutical industry( ex. ICH, ICH Global Cooperation Group Members).
- Strengthening the regulatory capacity of governments to manage, assess, and regulate drug development ( such as providing information, tools, training & exchange programs).
References:
- https://clinicaltrials.gov/
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents
- https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-classification-database

