




What Is Post-Marketing Surveillance?
Post-marketing surveillance (PMS) is the ongoing assessment of the safety, quality, and effectiveness of drugs, biological products, or medical devices after they have been authorized for use by the public. It is sometimes referred to as Phase IV post marketing surveillance, and it is intended to discover rare, long-term, or new side effects that may not occur in clinical trials prior to authorization.
The post marketing surveillance process is a vital and dynamic process in the lifecycle of a product; it forms real time data, assesses benefit-risk balance and regulatory action if necessary. As such, PMS acts as a link between clinical experimentation and safety for public health.
Although clinical trials provide useful information regarding the efficacy and safety of new drugs, biological products, and medical devices, the information captured through them is not all-encompassing. Limited patient populations, controlled and strict conditions treatment regimens, adherence protocols, limited duration and indications for use, and continuous monitoring of participants by healthcare professionals is not reflective of the real-world scenario once the product is approved and on the market. Post-marketing surveillance (PMS) studies refer to the monitoring of drugs once they reach the market after clinical trials and help to capture previously undetected or unexpected safety concerns that can threaten patient health. Additionally, PMS studies can also help positive effects of a drug in conditions not studied in clinical trials. Historical events such as the thalidomide tragedy that led to birth defects in children of women taking the drug for sleeplessness, anxiety, and morning sickness have supported the use of PMS studies. Therefore, it is necessary for manufacturers and sponsors must develop a robust PMS program which is also required as part of Phase IV studies by regulatory authorities and a component of risk management plan.
PMS clinical studies result in appropriate action to improve patient safety which includes changes in product labelling (dose regimen, interactions, adverse effects), product withdrawal from the market, or restricted use in certain patient populations (paediatrics or geriatrics). These changes can be instituted voluntarily by drug companies or forced upon by regulatory authorities, surveillance organizations, consumer advocacy groups, and specialist commentators. However, the overarching goal of all PMS studies is to ensure the safety of drugs and medical devices and to ensure that appropriate actions are undertaken if the risk of continued use outweighs the benefit.
Post-Marketing Surveillance in Pharmacovigilance and Regulatory Affairs
Post-Marketing Surveillance in pharmacovigilance and regulatory affairs. After a drug product is marketed, PMS should continue to monitor the safety, efficacy, and quality of the drug. While pre-marketing clinical trials can be a rich source of data, the data collected is typically limited in scope, occurs in controlled settings, and is usually limited to a specific population. Post-marketing surveillance is critical because clinical trials involve limited populations, and rare adverse events often appear only after widespread real-world use.Once a drug product is marketed, PMS ensures that the product is monitored in the “real world,” or in a wide-range of exposure and larger populations, and extends over longer periods of time.
Through the surveillance of the drug in a “real world” manner, PMS continuously monitors drugs product characteristics and monitors for new or rare adverse drug reactions (ADRs), long-term effects, and possible drug-drug interactions that may not have been seen during clinical development. The data collected is from multiple “real-world” data, such as spontaneous reporting systems, prescription event monitors, patient registries, and observational studies. These reports are important for both manufacturers and regulators to continually re-evaluate and assess a product’s benefit-risk profile.
PMS is also relevant to regulators as part of their mandated pharmacovigilance programs, such as those mandated by FDA (in the U.S.), EMA (in Europe), and CDSCO (in India). PMS provides evidence (or signal) generation in making regulatory decisions, such as whether to request a label change, restrict use, or withdraw a product from the market. PMS also ensures a returned adherence to Good Pharmacovigilance Practices (GVP) and improves public confidence by providing transparency and communication.
Additionally, PMS data informs regulatory documents, namely Periodic Safety Update Reports (PSURs) and Periodic Benefit-Risk Evaluation Reports (PBRERs), which are essential aspects of ongoing regulatory submissions and lifecycle management plans. By bridging clinical practice to regulatory framework, PMS supports continuous safety assessments, appropriate risk mitigation, and informed policies.
Phrased differently, Post-Marketing Surveillance is a continued collaboration process that supports patient safety, improves evidence-based decision making and enables regulatory compliance in the product lifecycle.
The various Post Marketing surveillance strategies are as follows
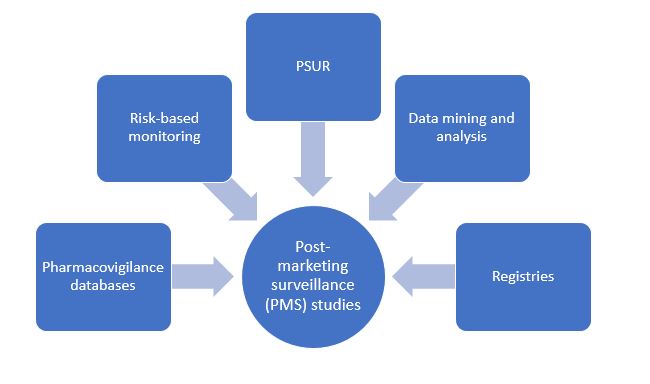
Traditional Post-Marketing Surveillance Strategies
Pharmacovigilance databases and programs
The FDA Adverse Event Reporting System (FAERS) is a database that contains adverse event and medication error reports, and product quality complaints that are voluntarily reported to the FDA by healthcare professionals or consumers. Clinical reviewers at the FDA evaluate and analyse the reports sent and make recommendations on the safety of the product. MedWatch is FDA’s program for mandatory and voluntary reporting of prescription medications, over-the-counter (OTC) products, and biologicals, and medical devices to the FAERS database and is responsible for the publication of safety alerts for FDA-regulated products. The FDA’s Sentinel initiative is an active post-market surveillance program that analyses safety data from real-world sources such as electronic health records (EHRs), insurance claims, registries, and pharmacy records. The Sentil system involves the contribution of several organizations that analyse the data and make decisions on action to be taken which can involve investigations and regulatory updates. In India, the Pharmacovigilance Program of India (PvPI) which is an Indian government organization is responsible for the identification of safety concerns and appropriate responses to protect patient health.
Risk-based monitoring
It is important to develop a risk management plan commensurate with the risk category of a drug or medical device throughout the product lifecycle to identify and remedy potential safety risks. This strategy involves a continuous collection of safety information, evaluation of risk-benefit profile, and timely and comprehensive risk communication to healthcare professionals and consumers regarding safety issues.
Data mining and analysis
Data mining and statistical analysis techniques are a crucial part of PMS clinical studies as pharmacovigilance databases contain data in different formats and standards from sources such as clinical trials or spontaneous reports. Decoding, cleaning, and normalization of data sets is important to extract meaningful information about safety concerns. Disproportionality analysis, Bayesian data mining, and machine learning (ML) are used to analyses safety signals and their severity and are performed on an ongoing basis to ensure product safety throughout the lifecycle.
Periodic Safety Update Report (PSUR)
PSURs are pharmacovigilance documents that provide an evaluation of risk-benefit balance of a pharmaceutical product/medical device at defined time points after its authorization or approval by regulatory agencies. It contains detailed information on the adverse events, patient populations, relevant literature, and previous regulatory actions. They allow for timely detection of adverse events and allows for the development of risk mitigation strategies.
Registry programs
Disease and product registries provide surveillance for a broad patient population which is inclusive of gender, age, and comorbidities and provides information of prescription patterns and off-label uses and serve as an important source of real-world data (RWD).
Thus, PMS studies are as important as clinical trials which are used to obtain regulatory approval for new drugs and medical devices. Integrating data from various types of PMS studies is critical and is monitoring the safety of the drug on a continuous basis to allow for prompt regulatory actions that can help prevent major adverse events that can jeopardize patient health.
Advanced & Emerging Post-Marketing Surveillance Strategies
In addition to traditional pharmacovigilance approaches, modern post-marketing surveillance strategies increasingly leverage digital technologies and real-world data to enhance drug safety monitoring and regulatory compliance.
AI and Machine Learning for Signal Detection
Artificial intelligence and machine learning algorithms are transforming pharmacovigilance by automating adverse event case processing, literature monitoring, and safety signal detection. These technologies enable early identification of potential safety risks and improve decision-making.
Real-World Evidence (RWE) Integration
Real-world evidence from electronic health records, insurance claims, patient registries, and wearable devices is increasingly used to complement traditional post-marketing studies. RWE provides insights into long-term safety, treatment effectiveness, and population-specific outcomes.
Digital Patient Monitoring and Remote Data Collection
Mobile health applications, wearable devices, and telemedicine platforms enable continuous monitoring of patient outcomes and adverse events in real-world settings. Digital monitoring improves patient engagement and data accuracy.
Social Media and Patient-Reported Outcome Surveillance
Monitoring social media platforms and patient forums helps detect emerging safety signals and patient-reported adverse events that may not be captured through traditional reporting systems.
Personalized Pharmacovigilance
Precision medicine approaches tailor safety monitoring based on genetic, demographic, and clinical risk factors. Personalized pharmacovigilance enhances risk assessment and improves patient safety.
Global Safety Data Collaboration
Pharmaceutical companies and regulatory agencies are increasingly collaborating and sharing safety data across global databases to accelerate signal detection and improve public health outcomes.
What Is Post-Marketing Surveillance of Drugs
Post-marketing surveillance of drugs refers to the systematic monitoring of pharmaceutical products after they have received marketing authorization and are widely used in clinical practice. The primary objective is to identify rare, delayed, or population-specific adverse drug reactions (ADRs) and to continuously evaluate the drug’s benefit–risk profile in real-world conditions.
Unlike pre-approval clinical trials, which involve limited patient populations and controlled protocols, post-marketing surveillance of drugs captures safety and effectiveness data from broader and more diverse populations over extended periods. This ongoing monitoring helps regulatory authorities and manufacturers ensure that therapeutic benefits continue to outweigh potential risks throughout the drug’s lifecycle.
Post-Marketing Surveillance of Medical Devices
Post-marketing surveillance of medical devices involves the continuous assessment of device performance, safety, and quality after commercialization. Medical devices are often used in complex clinical environments and across heterogeneous patient populations, making real-world monitoring essential for detecting device malfunctions, usability issues, and long-term safety concerns.
Key objectives include:
- Detecting device-related adverse events and malfunctions
- Evaluating long-term performance and durability
- Monitoring off-label use and real-world clinical outcomes
- Ensuring compliance with regulatory requirements (FDA, MDR, CDSCO, etc.)
Post-marketing surveillance of medical devices plays a critical role in preventing device recalls, improving device design, and ensuring patient safety in routine clinical practice.
Post-Marketing Surveillance Study
A post-marketing surveillance study is a structured research activity conducted after a product is approved to generate real-world evidence on safety, effectiveness, and utilization patterns. These studies are often referred to as Phase IV clinical studies and are conducted as part of regulatory commitments or risk management plans.
Common types of PMS studies include:
- Phase IV interventional clinical trials
- Observational cohort and case-control studies
- Patient registries and disease registries
- Prescription event monitoring studies
- Real-world evidence studies using EHRs and claims databases
Post-marketing surveillance studies provide scientifically robust evidence that supports regulatory decision-making, label updates, and pharmacovigilance signal validation.
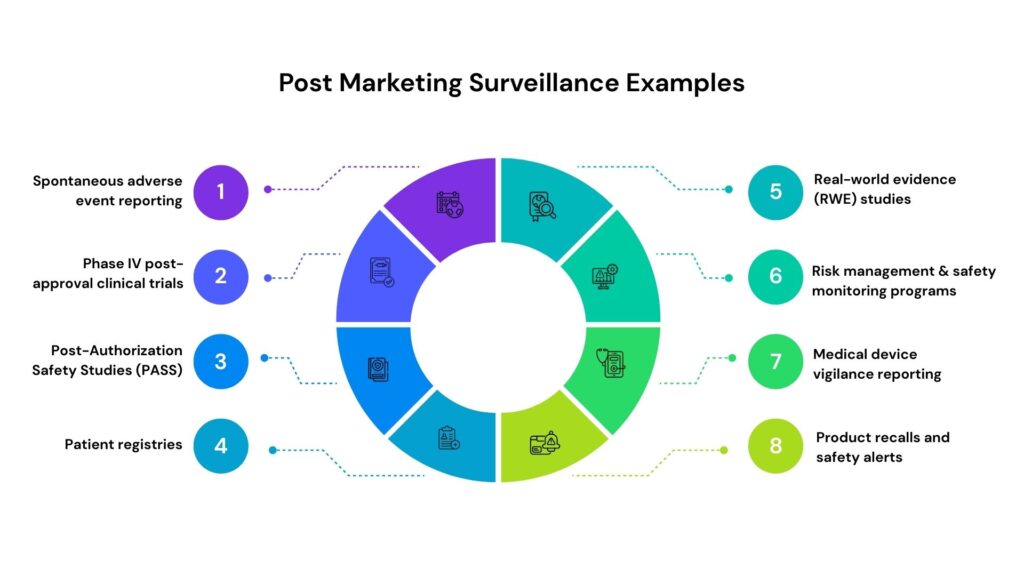
Post-Marketing Surveillance Examples
Post-marketing surveillance (PMS) is a critical pharmacovigilance and regulatory compliance activity conducted after a drug, biologic, medical device, or nutraceutical is approved and marketed. It enables continuous monitoring of real-world safety, effectiveness, and long-term risk-benefit profiles across diverse populations. Through structured and unstructured data collection, PMS helps identify rare adverse events, validate clinical trial findings in real-world settings, and support regulatory commitments, thereby strengthening patient safety and product lifecycle management.
Post-Marketing Surveillance Mechanisms and Techniques
Post-marketing surveillance mechanisms are important aspects of pharmacovigilance aimed at continually monitoring medicine safety, effectiveness, and other characteristics after marketing authorization and release into the market. The surveillance mechanisms involve systematic collection, analyses, and interpretation of data involving adverse drug reactions (ADRs), product quality concerns, and long-term real-world therapeutic effects. While there are many PMS methods, the most common mechanisms include spontaneous reporting systems, patient registries, observational studies, periodic safety update reports (PSURs), and active surveillance studies. Regulatory authorities and pharmaceutical companies work collaboratively to identify new safety signals, evaluate the benefit-risk profile, and undertake risk minimization measures such as labelling revisions or product withdrawals when needed. By defining and executing PSM mechanisms, PMS is essential in providing protection to the public, improving medicine safety, and ensuring confidence in the healthcare system.
Post-Marketing Surveillance Techniques as follows
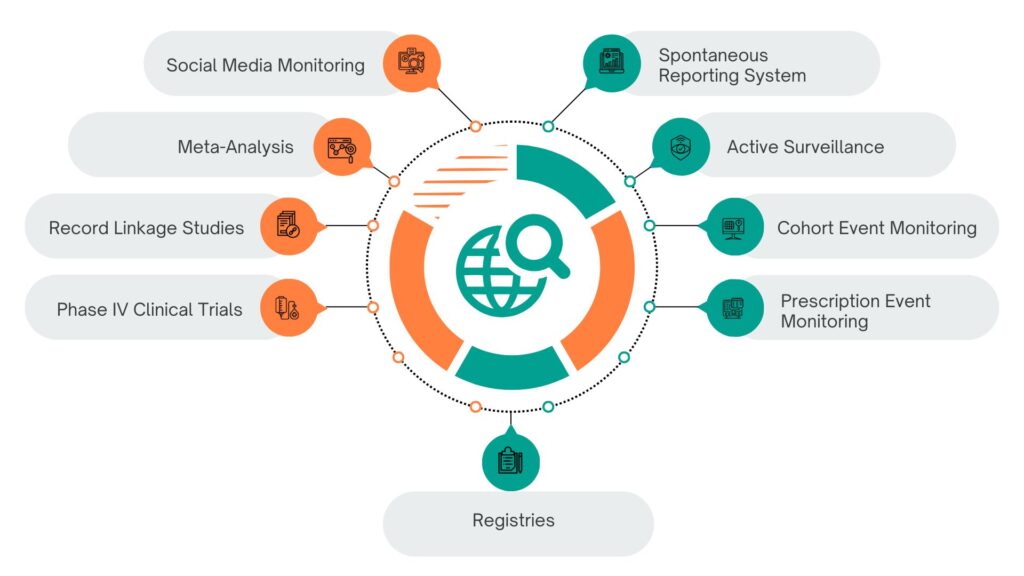
1. Spontaneous Reporting System (SRS)
This is one of the more common methodologies where healthcare professionals, pharmaceutical companies, or patients voluntarily report suspected adverse drug reactions (ADRs) to national or international regulatory authorities (e.g., MedWatch from the FDA, VigiBase from WHO). Each system of spontaneous reporting helps to identify safety signals earlier.
2. Active Surveillance
Active surveillance collects data through proactive methods including registries, follow-up studies, and electronic health record (EHR) monitoring, to identify adverse events. Active surveillance aims to issue real-world evidence by even collecting data on patient exposures to aid in cleaning the use of medications within clinical practice.
3. Cohort Event Monitoring (CEM)
Cohort Event Monitoring (CEM) involves monitoring a cohort of patients exposed to a specific drug over time, recording each adverse event in a systematic method. Cohort event monitoring makes it possible to estimate incidence rates and risk profiles of specific drug exposures in real-world environments.
4. Prescription Event Monitoring (PEM)
Prescription Event Monitoring (PEM) includes monitoring large populations prescribed a new medicine, collecting data directly from prescribers or pharmacies. This methodology can ultimately provide insight into utilization, safety, and effectiveness of medications within everyday clinical practice.
5. Phase IV Clinical Trials
Also referred to as post-marketing studies, these studies are performed following the approval of a drug to provide information about its longer-term safety, ideal usage, and its effectiveness in comparison to other treatments.
6. Record Linkage Studies
These studies utilize linked data from diverse healthcare databases (for instance, hospital records, prescription records) to discover associations between drug exposure and adverse events, thus enhancing the reliability of signal detection.
7. Meta-Analysis and Literature Review
Analysis of published data and analysis of multiple studies together is useful for identifying unusual adverse reactions and verifying safety signals identified using other methods.
8. Social Media and Digital Monitoring
Developing pharmacoepidemiological tools examine Twitter, Facebook, and the rest of the digital world for discussion, mentions, and post of this health related adverse events involving drugs, providing a preliminary early warning system from real users.
Post-Marketing Surveillance Report
A post-marketing surveillance report is a regulatory document that summarizes safety data, adverse event trends, benefit–risk evaluations, and risk mitigation activities for an approved product. These reports are submitted periodically to regulatory authorities to ensure ongoing compliance and transparency.
Common PMS regulatory reports include:
- Periodic Safety Update Reports (PSURs)
- Periodic Benefit-Risk Evaluation Reports (PBRERs)
- Risk Management Plans (RMPs)
- Development Safety Update Reports (DSURs)
Post-marketing surveillance reports serve as a critical communication tool between pharmaceutical companies and regulators, ensuring continuous monitoring and proactive risk management throughout the product lifecycle.
What is the post marketing surveillance process?
The post-marketing surveillance process is the ongoing monitoring of safety and effectiveness for approved drugs, biologics, or medical devices once they are available on the market. Post-marketing surveillance involves the collection of real world data from numerous sources such as: adverse event reports, patient registries, electronic health records and the literature. The data is validated, coded and analyzed to identify any safety signals or new risks. Once a risk has been identified, a risk evaluation is performed against the therapeutic benefits, noted in a risk management plan, and communicated to regulatory authorities, healthcare providers, and patients (when appropriate). Post-marketing surveillance of drugs, biologics and medical devices reflects an ongoing commitment to monitor safety signals to ensure that product labels are updated in a timely manner, and that actions are taken when necessary to mitigate risk to patients.
Final Remarks
Post-Marketing Surveillance (PMS) is a significant component in verifying the continued safety, efficacy, and quality of pharmaceutical products and medical devices, after they are on the marketplace. While pre-marketing clinical trials offer valuable information about products under controlled conditions, PMS generates real-life, ongoing, and diverse monitoring of products in routine clinical practice and patient populations and for longer periods of time. Monitoring approaches, such as spontaneous reporting systems, active surveillance, registries, cohort-event monitoring, and data mining, facilitate the timely detection of rare adverse drug reactions (ADRs) or long-term ADRs, which may be undetectable during clinical development.
PMS is fundamental to evidence-based decision making, risk management, and policy development from both a regulatory and pharmacovigilance standpoint. PMS can support decisions regarding timely interventions, e.g., labelling changes, restricted use, or withdrawing a product entirely from the market, thereby protecting public health. PMS is an important regulatory means of supporting compliance, transparency, and public trust in healthcare systems.
A proactive and robust PMS framework is the foundation for transforming monitoring of post-marketing products to safety monitoring back into a complete cycle of ongoing benefit/risk evaluation for the life of the drug or device. This allows for ongoing therapeutic advantages to be realized, while minimizing potential risks, over the course of the medicinal product and medical device life span.
FAQS
What is the post marketing surveillance definition?
The post marketing surveillance definition is the continuous monitoring of a drug, biologic, or medical device after approval to identify adverse events, evaluate real-world safety, and assess long-term effectiveness.
What is the post marketing surveillance stage?
The post marketing surveillance stage begins after regulatory approval (Phase IV) and focuses on monitoring real-world safety, effectiveness, and risk–benefit balance throughout the product lifecycle.
What is phase 4 post marketing surveillance ?
Phase 4 post marketing surveillance refers to post-approval studies and real-world monitoring conducted after a product is marketed to detect rare adverse reactions, long-term effects, and optimize clinical use.
What are the objectives of post-marketing surveillance?
The objectives of post-marketing surveillance are to detect rare adverse events, monitor long-term safety, evaluate real-world effectiveness, and maintain a favorable benefit–risk profile of marketed products.
What are the key data sources for post-marketing surveillance?
The key data sources for post-marketing surveillance include spontaneous adverse event reports, electronic health records (EHRs), insurance claims databases, patient registries, observational studies, and real-world evidence (RWE) platforms.
Who is responsible for post-marketing surveillance?
Responsibility for post-marketing surveillance lies with pharmaceutical companies, medical device manufacturers, regulatory authorities such as FDA, EMA, and CDSCO, healthcare professionals, and pharmacovigilance organizations.
What actions can result from post-marketing surveillance findings?
Actions resulting from post-marketing surveillance findings may include label changes, safety warnings, restricted use, risk mitigation measures, product recalls, or market withdrawal.
How long does post-marketing surveillance continue?
Post-marketing surveillance continues throughout the entire product lifecycle, as long as the drug or medical device remains on the market.
References
- Postmarketing Surveillance – an overview | ScienceDirect Topics
- Guidance for post-market surveillance and market surveillance of medical devices, including in vitro diagnostics (who.int)
- FDA’s Sentinel Initiative – Background | FDA
- MedWatch: The FDA Safety Information and Adverse Event Reporting Program | FDA
- Pharmacovigilance Programme of India (ipc.gov.in)
- Post-marketing Drug Safety Evaluation using Data Mining Based on FAERS – PMC (nih.gov)
- Use of Registries in Product Safety Assessment – Registries for Evaluating Patient Outcomes – NCBI Bookshelf (nih.gov)
